25/07/2025
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
La FDA advierte sobre el riesgo grave de complicaciones por calor asociado al parche antinaúseas transdérmico de escopolamina (Transderm Scōp)
(FDA issues warning about serious risk of heat-related complications with the antinausea patch scopolamine transdermal system transderm scōp)
Worst Pills Best Pills, 14 de julio de 2025.
https://www.worstpills.org/e-alerts/view/147
Traducido por Salud y Fármacos
Tags: grave riesgo de complicaciones relacionadas con transderm scōp, el parche antinaúseas transderm scōp puede causar graves complicaciones, muerte en niños y adultos mayores asociada al uso de transderm scōp
El 18 de junio de 2025, la FDA emitió una advertencia de seguridad para quienes usan el sistema transdérmico de escopolamina (transderm scop), un parche que se utiliza para tratar las náuseas y los vómitos, pues pueden experimentar complicaciones relacionadas con el calor [1]. El parche puede aumentar la temperatura corporal y disminuir la sudoración, lo que puede provocar complicaciones relacionadas con el calor, como confusión, pérdida del conocimiento, hospitalización o, en algunos casos, incluso la muerte.
La FDA recomienda que cualquier persona, especialmente los menores de 17 años y los adultos mayores de 60 años, que experimente síntomas de hipertermia como temperatura corporal elevada o disminución de la sudoración se retire el parche inmediatamente. Estas personas también deben evitar el uso de fuentes de calor externas como mantas térmicas, y deben mantenerse alejados de ambientes cálidos.
El sistema transdérmico de escopolamina, aprobado inicialmente en 1979, es un parche que se utiliza en adultos para tratar las náuseas y los vómitos asociados con el mareo por movimiento y la recuperación de la anestesia [2]. El parche para tratar las náuseas funciona liberando escopolamina, un anticolinérgico que bloquea las señales cerebrales responsables de las náuseas y los vómitos. El parche se aplica detrás de la oreja y libera un miligramo de escopolamina, cuyo efecto antiemético (evitar el vómito), comienza en cuatro horas y dura hasta tres días.
Las personas con glaucoma de ángulo cerrado o hipersensibilidad a la escopolamina no deben utilizar el parche. Aunque no está aprobado para su uso en niños, el sistema transdérmico de escopolamina se ha recetado ocasionalmente fuera de indicación para tratar la salivación excesiva en niños con parálisis cerebral u otros trastornos neurológicos [3].
La advertencia de seguridad sobre la escopolamina transdérmica se ha actualizado para añadir el riesgo de hipertermia a la información para la prescripción y al prospecto para el paciente. Los agentes anticolinérgicos como la escopolamina pueden elevar la temperatura corporal central y disminuir la sudoración. Estos efectos pueden agravarse por la exposición a fuentes externas de calor o en entornos con altas temperaturas. Estos síntomas, si no se detectan o no se tratan, pueden provocar complicaciones graves. En la mayoría de los casos la hipertermia se presenta dentro de las 72 horas posteriores a la aplicación. Incluso después de retirar el parche, los síntomas de abstinencia como mareos, dolor de cabeza y náuseas, pueden persistir durante varios días, ya que el medicamento absorbido permanece en el organismo [4].
La advertencia de la FDA se basa en 13 casos de hipertermia reportados a nivel global en pacientes que usan el parche para tratar las náuseas, incluyendo siete en EE UU. De estos casos, ocho involucraron a niños menores de 17 años y cuatro a adultos mayores de 60 años. Cuatro casos resultaron en hospitalización y dos pacientes murieron: uno era un niño y el otro un adulto mayor.
“En base al funcionamiento del medicamento, incluyendo su capacidad para atravesar la barrera hematoencefálica, una membrana que impide que sustancias potencialmente dañinas de la sangre lleguen al cerebro, la FDA determinó que existe evidencia razonable de asociación causal entre los parches de escopolamina y la hipertermia” [4].
El Grupo de Investigación en Salud de Public Citizen recomienda que, si utiliza el sistema transdérmico de escopolamina, esté atento a cualquier signo de calor excesivo o alteración de la temperatura, y que si presenta síntomas se retire el parche inmediatamente y se ponga en contacto con su médico.
Para consultar la alerta de la FDA, visite el siguiente enlace: https://www.fda.gov/media/187121/download
Informe cualquier evento adverso asociado con la escopolamina transdérmica (Transderm Scōp) al programa MedWatch de la FDA llamando al 1-888-463-6332 o visitando https://www.accessdata.fda.gov/scripts/medwatch/
Referencias
Petro del lado de la industria farmacéutica transnacional
Bernardo Useche Aldana
La Tribuna, 13 de julio 2025
https://latribunacolombia.co/petro-del-lado-de-la-industria-farmaceutica-transnacional/
Tags: Acceso universal a medicamentos, justicia sanitaria global, reformar sistema de patentes, Propiedad intelectual y salud pública, Industria farmacéutica transnacional, Comités de ética en investigación, Ensayos clínicos en América Latina.
La industria farmacéutica transnacional, con lineamientos del Banco Mundial, impulsa un plan para convertir a Colombia en centro regional de ensayos clínicos en América Latina. Declaraciones empresariales muestran que se busca redirigir recursos públicos, reformar los comités de ética y ampliar la presencia de multinacionales en el sistema de salud.
La Ley 100 introdujo como actores principales en el sistema a las EPS y el modelo de aseguramiento social en salud. En el panorama que se avecina luego del desbarajuste ocasionado con la política implementada por el presidente Petro y sus ministros de salud, las multinacionales farmacéuticas representadas por la Asociación de Laboratorios Farmacéuticos de Investigación y Desarrollo, AFIDRO, buscan jugar un rol preponderante en el nuevo sistema de salud que termine por configurarse.
El objetivo va más allá: “consolidar a Colombia como un hub regional de investigación” en América Latina. El interés de la industria farmacéutica en este empeño se basa en el hecho de que el mercado farmacéutico latinoamericano es uno de los de mayor crecimiento en el mundo con una población creciente de 660 millones adultos mayores. Interés incentivado por las recientes órdenes ejecutivas expedidas por el presidente Trump con el propósito de acelerar la producción de nuevas medicinas y disminuir los precios de los medicamentos en Estados Unidos, lo que obliga a las grandes farmacéuticas a aumentar los precios y los mercados en el exterior (Executive Order 14273 of April 15, 2025; Executive Order 14293 of May 5, 2025 – Executive Order 14297 of May 12, 2025).
El papel del Banco Mundial
Si en 1993 la reforma neoliberal de salud en Colombia en beneficio de las compañías aseguradoras en salud fue diseñada por el Banco Mundial, hoy, el Banco ha definido también las líneas centrales de la política de participación de las gran (https://openknowledge.worldbank.org/entities/publication/e5fae256-f0c0-5400-9c14-b80387e182c8) industria farmacéutica en los sistemas de salud en América Latina y el Caribe:
Las orientaciones del Banco Mundial se han materializado en una estrategia formulada por la Federación Latinoamericana de la Industria Farmacéutica (https://afidro.org/wp-content/uploads/2025/06/FIFARMA-Investigacion_Clinica_en_Latinoamerica_2.pdf), organización que agrupa “empresas farmacéuticas multinacionales líderes” y que actualmente está bajo la dirección de Yaneth Giha, exdirectora de AFIDRO y Ministra de Educación en el segundo gobierno de Juan Manuel Santos.
Aseguradoras y farmacéuticas
El propósito de los grandes laboratorios y compañías farmacéuticas es en primer lugar, articularse a los sistemas de salud y elevar las ventas de sus medicamentos y tecnologías con cargo a los recursos del Estado. Otro documento de la federación es claro al respecto: “El llamado que hacemos desde FIFARMA (…) es que los gobiernos de la región pongan en marcha planes de incremento en sus presupuestos para el sector salud”.
Aseguradoras y farmacéuticas coinciden en que se aumente el presupuesto general en salud, pero tienen entre sí una contradicción económica. A mayor gasto en medicamentos menor presupuesto disponible para el aseguramiento. En la superficie se clama por el aumento del financiamiento público de la salud y de la UPC para responder a las tutelas, quejas y reclamos de la población, lo cual tiene sentido en la crisis actual de la salud, profundizada en el presente gobierno.
En el fondo, las casas farmacéuticas se aprestan a competir con las aseguradoras por un mayor monto de los recursos del Estado. Esto implica una disputa por los servicios y tecnologías que se incluyan en el plan de beneficios y que se contraten con hospitales y clínicas. A propósito, un riguroso estudio reciente del sistema de salud de Taiwán (https://www.sciencedirect.com/science/article/pii/S0929664624004790?via%3Dihub) encontró que a medida que, con recursos del Estado, aumentaron las ganancias de las farmacéuticas, más se afectó la estabilidad financiera de los hospitales.
Más ensayos clínicos en Latinoamérica, parte de la estrategia global de frenar a China
En este contexto, las multinacionales farmacéuticas han lanzado un plan para convertir a Colombia en tierra fértil que produzca un “crecimiento exponencial” de los ensayos clínicos con los que la industria crea productos farmacéuticos de “innovación” cuyas patentes garantizan altísimos precios y monopolios en el mercado.
No extraña entonces que el Dr. Ignacio Gaitán, presidente actual de AFIDRO, haya declarado que: “América Latina podría cuadruplicar su participación en estudios clínicos”. Este plan de expansión de los estudios clínicos en los que se evalúa si un nuevo medicamento tiene efecto preventivo o curativo (eficacia), y si no produce efectos adversos de consideración para la salud de los participantes (seguridad), se encuadra en el reconocimiento por parte de la industria de que “la investigación clínica es una actividad altamente competitiva, que se desarrolla en un entorno global”.
En particular, para las multinacionales norteamericanas incrementar el número de ensayos clínicos en Latinoamérica se inscribe en la estrategia global de la industria de frenar el avance de China en la Investigación y Desarrollo (I & D) de productos farmacéuticos innovadores.
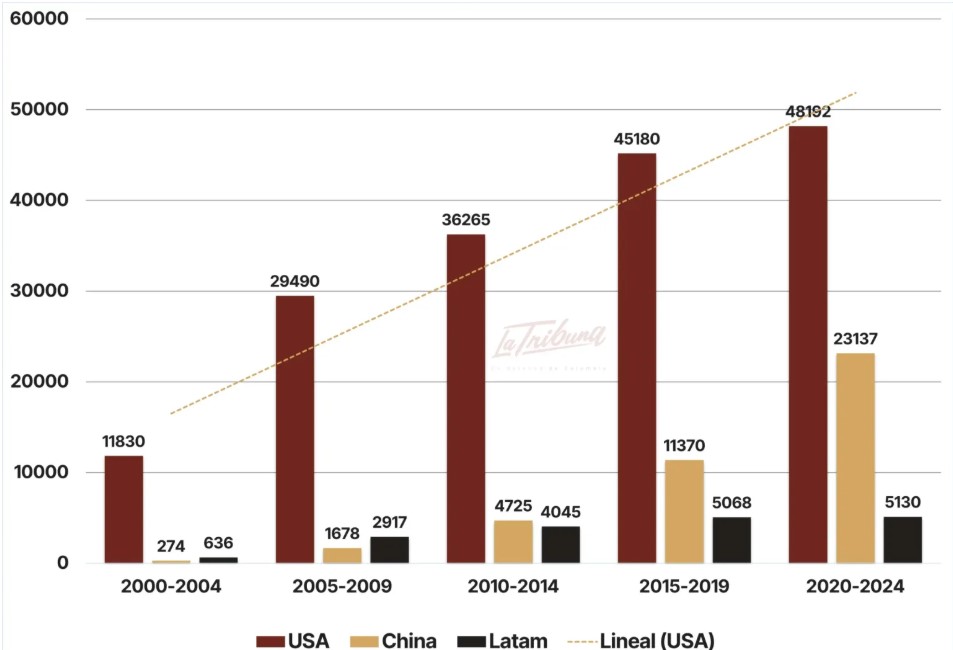
En el período 2000-2004, el número de ensayos clínicos iniciados en China (274) fue 40 veces menor que los estudios emprendidos por Estados Unidos (11.830). En el período 2020-2024, Estados Unidos inició 48.192 ensayos clínicos, solo dos veces el número de los estudios iniciados por China (23.137). De continuar su ritmo de crecimiento, China será en corto tiempo el país a la vanguardia en la investigación farmacéutica, lo que tendrá un gran impacto en el mercado global a mediano plazo.
Las grandes farmacéuticas consideran que Colombia y otros países latinoamericanos ofrecen condiciones muy favorables para su política de competir con China en la investigación de nuevos productos. El gasto público en salud crece, la población es étnica y racialmente diversa, el sistema de salud cubre la casi totalidad de la población, los costos de los ensayos clínicos son muy inferiores a los realizados en Europa o Norteamérica.
El gobierno de Gustavo Petro ha visto con buenos ojos el posicionamiento de las multinacionales en el país. Un documento del Ministerio de Salud de 2023 lo plantea: “En Colombia, la investigación clínica patrocinada por la industria farmacéutica ha ido avanzando en los últimos 30 años, posicionándose en el cuarto lugar en Latinoamérica y siendo uno de los países de gran interés por parte de la industria gracias a la infraestructura, calidad de los profesionales de la salud y cobertura del sistema de salud”.
En consonancia con el Ministerio, el mes de mayo pasado, María Fernanda Velasco, directora médica de Pfizer Colombia informó: “Pfizer ha invertido más de 524.5 millones de dólares en investigación clínica desde 2020, posicionando la región como un punto estratégico para la validación científica de tratamientos innovadores para enfermedades complejas que afectan a muchas personas”. La Doctora Velasco agregó que Pfizer cuenta con más de 300 centros de investigación en Brasil y más de 250 en otros países de la región, incluyendo a Colombia.
El papel de los Comités de Ética en Investigación
Ahora, multiplicar de manera expedita los ensayos clínicos requiere que los estudios obtengan rápida aprobación por Comités de Ética de la Investigación (CEI). Estos comités surgieron luego de que una comisión constatara los delitos atroces cometidos durante experimentos médicos con prisioneros en campos de concentración Nazis y otros casos notorios de abuso (Ver Tabla 1).
Posteriormente, en distintos momentos de la historia se ha documentado la existencia de condenables abusos con personas incluidas en diversos estudios clínicos (ver Tabla 2). Se hizo entonces necesario formular criterios y normas éticas y crear los CEI para garantizar la protección de las personas o pacientes participantes en este tipo de estudios.
Las compañías transnacionales de productos farmacéuticos consideran que el funcionamiento de los CEI es lento, que los criterios y normas éticas reguladoras de la investigación clínica se deben unificar internacionalmente y en últimas, que la aprobación de los ensayos debería hacerse en el país “por parte de un único comité”.
La propuesta se orienta a que los CEI sean reemplazados por Organizaciones de Investigación por Contrato o CRO por sus iniciales en inglés (Contract Research Organizations), compañías privadas afines a los intereses de la gran industria farmacéutica.
Los CEI no están exentos de problemas. Un estudio en ocho países latinoamericanos que hicimos bajo la dirección de Nuria Homedes y Antonio Ugalde encontró que, tanto en Colombia como en la región, los comités adolecen de falencias que debilitan su función protectora de los participantes, están expuestos a conflictos de interés con los patrocinadores y tienden a no dar prioridad a proyectos de investigación dirigidos a resolver las necesidades de salud de las poblaciones locales.
Sin embargo, un fuerte sistema de comités de ética independientes de la industria, centrado en la protección de los pacientes y en un conocimiento científico que ofrezca garantía de la seguridad y eficacia de los medicamentos investigados, podría dar mejores resultados que comités con protocolos orientados a favorecer la inversión de las multinacionales.
El dilema de Petro: del lado de las transnacionales o de la industria farmacéutica nacional
El gobierno a través del INVIMA convocó a la Primera Mesa Nacional de Investigación Clínica el pasado 11 de junio donde AFIDRO compartió su política de dar pasos adelante en posicionar al país como un hub en América Latina para el desarrollo de medicamentos que puedan ser patentados por las multinacionales farmacéuticas.
La gran industria farmacéutica debe mantener un lugar importante en el sistema de salud colombiano, pero la industria farmacéutica nacional debe fortalecerse. Las decisiones que se tomen sobre los CEI y las condiciones en que agilicen los trámites del INVIMA y la regularización de los ensayos clínicos van a definir el nivel de incidencia de las multinacionales en el sistema de salud colombiano.
Es una prioridad que se revise el capítulo 16 del TLC con Estados Unidos sobre propiedad intelectual. Es fundamental que la investigación clínica con recursos públicos conduzca a productos con patentes nacionales. Es el único camino para que Colombia se ubique en la senda de la seguridad y la soberanía farmacéutica.
El acceso a medicamentos es un derecho, no un privilegio
Salud y Fármacos
Tags: Acceso universal a medicamentos, justicia sanitaria global, reformar sistema de patentes, Propiedad intelectual y salud pública, Producción farmacéutica local, soberanía sanitaria en materia farmacéutica.
En mayo de 2025, la ciudad de Marrakech fue escenario de la Cumbre Mundial sobre Propiedad Intelectual y Acceso a los Medicamentos (Global Summit on Intellectual Property and Access to Medicines, GSIPA2M) [1], un encuentro que reunió a líderes comunitarios, expertos legales, académicos, funcionarios gubernamentales y activistas de todo el mundo para reflexionar sobre los 30 años del Acuerdo sobre los Aspectos de los Derechos de Propiedad Intelectual relacionados con el Comercio (ADPIC, TRIPS por su sigla en inglés).
El lema del evento, “TRIPS@30: The Access Crisis”, refleja el interés en hacer un análisis crítico de las consecuencias que este acuerdo internacional ha tenido sobre el acceso equitativo a medicamentos esenciales, especialmente en el Sur Global. El encuentro fue inaugurado por destacados referentes en el tema como Mohammed Zniber (ITPC, International Treatment Preparedness Coalition. MENA, Middle East and North Africa), el Dr. Houssine El Rhilani (UNAIDS, United Nations for Human Immunodeficiency Syndrome, Marruecos) y Solange Baptiste (ITPC Global), quienes expusieron con claridad la dimensión política del acceso a medicamentos. Denunciaron un modelo farmacéutico global centrado en el lucro, dependiente de los monopolios y estructurado en torno a patentes que obstaculizan el acceso a tratamientos que salvan vidas.
En palabras de Baptiste, “para millones, ‘acceso’ sigue siendo solo un eslogan. La innovación sin acceso no es progreso, es injusticia”. Durante los tres días del congreso se presentaron numerosos testimonios y análisis que revelaron el impacto devastador del sistema actual en países de ingresos bajos y medianos. Representantes de organizaciones comunitarias como la Campaña de Acción por el Tratamiento (Treatment Action Campaign,Sudáfrica), la Fundación GEP (Grupo Efecto Positivo, Argentina), Red de Personas Positivas de Delhi (DNP, Delhi Network of Positive People, India) y la Asociación Brasileña Interdisciplinaria del SIDA (ABIA, Associação Brasileira Interdisciplinar de AIDS, Brasil) compartieron experiencias de resistencia frente a barreras impuestas por la propiedad intelectual.
Se destacó el caso de Argentina, donde las oposiciones a solicitudes de patentes farmacéuticas impulsadas por la sociedad civil permitieron evitar monopolios injustificados y generaron un ahorro estimado de más de US$500 millones en gasto público en medicamentos en solo ocho años. Estos logros demostraron que, con organización, base legal y voluntad política, es posible modificar reglas establecidas que excluyen sistemáticamente a millones de personas.
Uno de los momentos más conmovedores del GSIPA2M fue el homenaje a la Dra. Hakima Himmich, referente en la lucha por el acceso a tratamientos para el VIH en el mundo árabe, fundadora de la primera asociación de VIH en Marruecos y médica especialista en enfermedades infecciosas. La Dra. Himmich ofreció un discurso titulado “Remembering to Resist”, donde repasó su trayectoria de lucha desde los años más oscuros de la epidemia. Relató cómo vio morir a sus pacientes sin acceso a tratamiento, cómo lideró campañas para eliminar impuestos a medicamentos y cómo forjó coaliciones nacionales e internacionales para garantizar atención gratuita. Su llamado final a las nuevas generaciones resonó profundamente en la audiencia: “No permitan que les digan que son demasiado jóvenes o demasiado radicales. Manténganse firmes en su indignación, esa es su brújula”.
El segundo día del encuentro se centró en la crisis de financiamiento internacional para la salud. Voces como la de Tracy Swan (Make Medicines Affordable) advirtieron que los recortes en la cooperación oficial al desarrollo están provocando desabastecimientos, interrupciones en tratamientos y una vulnerabilidad extrema en las comunidades afectadas por VIH, tuberculosis y hepatitis.
Andrew Hill, investigador de la Universidad de Liverpool, denunció el silencio de las grandes farmacéuticas que, tras obtener ganancias multimillonarias en los últimos años, se mantienen ajenas a las consecuencias humanas de esta crisis. Paralelamente, se analizaron los impactos de los tratados de libre comercio que imponen cláusulas ¨TRIPS-plus¨ y reducen el margen de acción de los países en desarrollo, afectando directamente su soberanía sanitaria.
También se presentaron experiencias gubernamentales exitosas que demostraron que es posible tomar decisiones políticas valientes orientadas a la salud pública. El caso más emblemático fue el de Colombia, que recibió un reconocimiento especial por emitir una licencia obligatoria sobre el medicamento dolutegravir, utilizado en el tratamiento del VIH. Esta medida fue resultado de la colaboración entre la sociedad civil, el Ministerio de Salud, el Ministerio de Industria y Comercio y la Superintendencia de Industria y Comercio, y demostró que la propiedad intelectual no puede estar por encima del derecho a la vida. Otros países como Egipto, Uganda, Moldavia, Honduras, Tailandia y Kirguistán también compartieron iniciativas que priorizan el acceso y la transparencia en los sistemas de compras públicas y patentes.
El tercer día del GSIPA2M estuvo dedicado al análisis de la producción local de medicamentos como estrategia clave para lograr soberanía sanitaria. Expertos de distintos países coincidieron en que, pese a los esfuerzos, muchos países del Sur Global siguen dependiendo de importaciones costosas y carecen de la infraestructura necesaria para producir incluso medicamentos básicos.
Se insistió en la necesidad de políticas públicas sostenidas, inversión estratégica y cooperación regional. Se propuso además un nuevo enfoque para la industria farmacéutica pública: descentralizada, basada en el conocimiento colectivo, capaz de utilizar licencias obligatorias y enfocada en el bienestar social antes que en la rentabilidad [1].
Las últimas sesiones abordaron temas interseccionales que profundizan la crisis del acceso: conflictos armados, desplazamientos forzados, catástrofes climáticas y regresiones democráticas. Panelistas de Ucrania, Brasil, Medio Oriente y Centroamérica relataron cómo estas crisis impactan directamente en los sistemas de salud, obstaculizan el trabajo de las organizaciones comunitarias y aumentan la represión sobre activistas. Se subrayó la importancia de la solidaridad transnacional y de una agenda de derechos humanos que incorpore salud, clima, género y migración como ejes inseparables.
El GSIPA2M concluyó con la presentación de una declaración política conjunta, redactada colectivamente por los participantes, que llama a una transformación estructural del sistema global de propiedad intelectual. El documento reafirma que el acceso a los medicamentos debe ser tratado como un derecho humano no negociable y exige una reforma urgente que anteponga la vida y la dignidad de las personas al interés económico de un puñado de corporaciones. La declaración fue acompañada por un compromiso explícito de continuar articulando acciones locales, regionales y globales en defensa de la salud pública.
A 30 años de la entrada en vigor del ADPIC el mensaje del GSIPA2M fue contundente: Mientras el acceso a tratamientos siga siendo un privilegio, la lucha será indispensable; frente a la exclusión, la respuesta será siempre la resistencia organizada.
Fuente Original
Bibliografía adicional relacionada
¿Más o mejores ensayos clínicos?
https://consultorsalud.com/mas-o-mejores-ensayos-clinicos/

Ante la avalancha de artículos que se acaban de publicar sobre la cantidad de ensayos clínicos que se llevan a cabo en Colombia y las estrategias para atraer más ensayos al país en aras de ¨dinamizar la economía¨, tal vez sea apropiado destacar otras opiniones de estudiosos del tema (1).
Colombia tuvo una destacada trayectoria en investigación biomédica e innovación pública en salud desde inicios del siglo XX. Con la transición al neoliberalismo y con la reforma del sistema de salud que introdujo la Ley 100, el país privilegió la adopción de tecnología foránea a finales del siglo XX. Desde entonces el énfasis ha sido atraer inversión extranjera, incluso con cesión gratuita de propiedad intelectual financiada con recursos públicos (2). Sin embargo, hasta donde sabemos, no se ha cuantificado la contribución neta de los estudios financiados por la industria farmacéutica en la economía colombiana, por lo que se desconoce cómo dicha política beneficia al país, más allá de los beneficios económicos que aporta a los que realizan los ensayos clínicos y a las instituciones que los albergan.
Colombia tiene un amplio marco normativo sobre la regulación de los Ensayos Clínicos (Res 8430/1993, Res 3823/1997, Res 1995/199, Res 2378/2008, las Guías emitidas por el INVIMA en 2015 y 2018, etc), pero al igual que sucede en otros países, estos instrumentos regulatorios no exigen que los responsables de diseñar, aprobar e implementar los ensayos clínicos tengan una formación ética sólida. Un certificado en Buenas Prácticas Clínicas resulta laxo como único requisito ético exigido a investigadores.
Estos instrumentos mencionan La Declaración de Helsinki y vale la pena resaltar que la última actualización de 2024 (3) logró avances importantes en la protección a los participantes de ensayos clínicos, especialmente en países en vías de desarrollo; sin embargo, no se suele verificar su aplicación.
Los contratos entre la industria farmacéutica y los investigadores y centros de investigación son secretos, por lo que se desconoce si encierran cláusulas que puedan comprometer la toma de decisiones del equipo investigador. También se ha documentado que no todos los miembros de los Comités de Ética de la Investigación (CEI), que son los que emiten conceptos de rechazo o favorabilidad de protocolos, están libres de conflictos de interés, sin que haya una supervisión efectiva del INVIMA (1).
La implementación de ensayos clínicos en América Latina se ve afectada por otros problemas críticos (4-9), por ejemplo la falta de independencia real de los CEl, la presión institucional y económica que se ejerce sobre los integrantes de los CEI e investigadores, la deficiente evaluación crítica de protocolos de investigación (en especial de los financiados con recursos privados) y la tendencia a priorizar la celeridad de aprobación por encima del análisis riesgo-beneficio o de estrategias para mejorar el acceso post-ensayo a nuevas tecnologías en salud eficaces (10).
Los participantes de ensayos clínicos han sido víctimas de múltiples atropellos y violaciones de derechos humanos fundamentales a lo largo de la historia, el estudio de la sífilis de Tuskegee, el estudio del cáncer de 1963 en el Hospital Judío de Enfermedades Crónicas de Brooklyn y el estudio de la hepatitis en Willowbrook hacen parte de la “santísima trinidad” de la bioética, según la historiadora Susan Reverby (11). Hoy persisten graves dificultades (12-16).
Por fortuna múltiples esfuerzos han logrado avances hacia el fortalecimiento de marcos regulatorios nacionales (17) e internacionales (18) que intentan proteger derechos fundamentales; sin embargo, persisten debilidades en la idoneidad de los consentimientos informados (uso de lenguaje técnico, falta de verificación real de comprensión por parte de los participantes, falta de detalles importantes para consentir de forma informada sobre los riesgos y probabilidades de éxito de los experimentos a los que se someten, los beneficios económicos para los investigadores y centros de investigación), la cobertura de eventos adversos de mediano y largo plazo (concluidos los ensayos clínicos) etc. No es infrecuente que el Estado termina asumiendo costos importantes por complicaciones y/o secuelas graves o discapacidad permanente de los participantes.
La falta de garantía en el acceso post-ensayo a los tratamientos que resultan eficaces, así como la aprobación de ensayos clínicos controlados con placebo cuando existen alternativas terapéuticas con beneficio demostrado, son otras falencias importantes. En este momento, las regulaciones de los ensayos clínicos no exigen que los patrocinadores de ensayos se comprometan a comercializar los productos que resulten exitosos en los países en donde se han realizado los ensayos clínicos, ni que lo hagan a precios asequibles.
Calificar los ensayos clínicos como una estrategia primordial para la ¨dinamización de la economía¨ debería estar antecedido por la priorización de protocolos de investigación que respondan a las necesidades en salud de la población y a la adherencia al método científico libre de conflictos de interés durante el desarrollo de esos ensayos; de forma que se garantice el menor riesgo para los participantes y el mayor beneficio posible para la población, incluido el acceso asequible al medicamento que los colombianos han contribuido a desarrollar.
Dicho esto, suena razonable y prudente fortalecer primero algunos tópicos críticos de calidad e integridad de la ciencia antes de aumentar el número de ensayos clínicos en países de bajos y medianos ingresos como Colombia.
Resumiendo, los principales tópicos a intervenir según académicos y expertos en el tema (1, 4-8) deben incluir el fortalecimiento de la soberanía en investigación con un aumento de la inversión estatal en I+D, la integración de postulados Helsinki 2024 (3) y CIOMS 2016 (18) y la generación de derechos de propiedad intelectual nacional cuando los ensayos clínicos se han realizado con recursos públicos.
Los expertos mencionan también que otorgar el acceso post ensayo a innovaciones en salud efectivas (especialmente cuando de ello depende la vida del participante) y crear un fondo público/privado que favorezca la gestión y reparación de daños causados a los participantes de ensayos clínicos serian mejoras a la protección de los derechos fundamentales a la salud y la vida.
En cuanto a los CEI es importante garantizar su independencia, recursos, remuneración y capacidad técnico-científica expedita para evaluar protocolos complejos con el mejor rigor metodológico y el menor sesgo posible. Establecer estándares éticos mínimos de obligatorio cumplimiento para todos los CEI e investigadores donde se prohíba los conflictos financieros para que la I+D estén al servicio de los problemas prioritarios de salud pública.
Hay que mejorar los repositorios públicos de los diferentes protocolos de investigación, con acceso oportuno y que incluya informes de seguimiento, enmiendas y decisiones éticas, así como establecer auditorías aleatorias por parte del Estado, agencias reguladoras y observatorios de la sociedad civil.
Aplicar los principios de sentido común (Common Sense) a ensayos clínicos oncológicos (19) apunta a generar resultados clínicamente significativos para los pacientes y transformadores como lo ha demostrado el imatinib y el rituximab.
Antes de ¨cuadruplicar los ensayos clínicos en Colombia¨, es urgente corregir las asimetrías de poder, los incentivos, los vacíos de regulación ética y minimizar los riesgos en el reclutamiento de participantes con vulnerabilidades mediadas por los determinantes sociales y comerciales de la salud en América Latina. En caso contrario se pueden ampliar las brechas de inequidad en el acceso a nuevas tecnologías en salud y multiplicar los riesgos y las falencias descritas.
Éticamente, se deben asegurar estándares robustos de consentimiento verdaderamente informado, justicia y transparencia para transformar el ecosistema actual a uno atractivo y confiable para la investigación en humanos y competitivo para países de bajos y medianos ingresos.
Referencias
Estándares de la FDA para la aprobación de fármacos para enfermedades mentales
Tags: aprobación regulatoria de fármacos psiquiátricos, fármacos aprobados por la FDA para enfermedades mentales, rechazo reiterado de psicofármacos antes de su aprobación, fármacos aprobados con menos de la mitad de los ensayos de eficacia positivos, eficacia de brexipiprazol, eficacia de pimavanserina, eficacia de gepirona, , desacuerdos entre funcionarios FDA y comités asesores de la fDA
Las enfermedades mentales son una de las principales causas de discapacidad a nivel mundial; sin embargo, el desarrollo de tratamientos farmacológicos eficaces continúa siendo limitado por el escaso conocimiento sobre la fisiopatología de los trastornos psiquiátricos y la falta de biomarcadores validados.
En este contexto, un estudio reciente analizó los ensayos clínicos que sirvieron de base para la aprobación de fármacos psiquiátricos por parte de la FDA entre 2013 y 2024, con el objetivo de evaluar la calidad de la evidencia utilizada en el proceso regulatorio [1].
El estudio incluyó 16 medicamentos recientemente aprobados para tratar diversas condiciones psiquiátricas, como depresión, esquizofrenia, trastorno bipolar, psicosis y Trastorno por Déficit de Atención con Hiperactividad. Se revisaron 73 ensayos clínicos, y según la FDA el 62% fueron positivos. No obstante, para tres de los medicamentos aprobados, menos de la mitad de los ensayos incluidos en la solicitud de comercialización fueron positivos: brexipiprazol (3 de 7 ensayos positivos), pimavanserina (1 de 4), y gepirona (2 de 12), y este último había sido previamente rechazado en tres ocasiones.
Por otra parte, la aprobación de pimavanserina se otorgó en contra de la recomendación inicial de los revisores médicos de la FDA, y tras la votación favorable del comité asesor.
Del total de ensayos revisados, el 63% (n=46) fueron considerados pivotales. Todos los ensayos pivotales fueron aleatorizados y de doble ciego. La mayoría (83%) fueron controlados con placebo y en un 15% adicional se emplearon comparadores activos además del placebo. Un 19% de los fármacos se aprobaron en base a los resultados de un solo ensayo pivotal.
Las escalas clínicas más utilizadas como criterio de valoración principal fueron la Escala de Síndrome Positivo y Negativo (PANSS), la Escala de Depresión de Montgomery-Asberg (MADRS) y la Escala de Depresión de Hamilton.
En cuanto a la población de los ensayos revisados, se observó una distribución similar entre mujeres (48,9%) y hombres (51,1%). Entre los ensayos que reportaron distribución por raza, fue mayoritariamente blanca (61,2%), raza negra (30%) y asiáticos (5,3%).
El estudio concluye que, aunque muchos de los medicamentos aprobados estaban respaldados por múltiples ensayos de fase 3, la calidad de la evidencia varió considerablemente entre los productos. En algunos casos, la aprobación se basó en una proporción relativamente baja de resultados positivos, lo que plantea interrogantes sobre la rigurosidad y consistencia de los estándares regulatorios.
Los autores destacan la necesidad de mejorar la transparencia y la aplicación uniforme de criterios regulatorios en el proceso de aprobación de medicamentos para enfermedades mentales, especialmente dada la sensibilidad del campo psiquiátrico, donde los tratamientos aprobados pueden tener efectos profundos sobre poblaciones vulnerables.
Fuente Original
Bibliografía adicional relacionada
Ecuador y América Latina debaten los conflictos de interés en políticas alimentarias
El lector se preguntará por qué Salud y Fármacos publica una nota sobre nutrición, y lo hacemos por dos razones. La primera es que con la creciente disponibilidad de productos para tratar la obesidad ha surgido una tendencia a medicalizar el sobrepeso, en lugar de invertir en políticas públicas que reviertan las tendencias nutricionales que han ocasionado el problema, y que responden a los intereses corporativos (entre otros la industria de la alimentación y las empresas de comida rápida).
La segunda razón es que tal como demostró Marion Nestlé en su libro Food Politics, las tácticas que utilizan la industria farmacéutica y de la alimentación para impedir la implementación de políticas públicas a favor de la salud son muy similares. Consecuentemente, los que trabajamos en el área de políticas farmacéuticas podemos aprender de las tácticas exitosas que implementan, o han implementado, otros grupos que promueven hábitos saludables, con frecuencia enfrentando a los poderes corporativos.
Todos estamos interesados en recordar a los tomadores de decisiones que las políticas públicas deben maximizar el bienestar de la población.
Quito, Ecuador. En el marco del conversatorio regional titulado “Una mirada al manejo de conflictos de interés en políticas alimentarias en Ecuador y la región”, realizado el pasado 29 de mayo y organizado por la Fundación de Fomento de la Investigación en Nutrición, Alimentación, Actividad Física y Salud Pública (SLAN, capítulo Ecuador) y el Comité de Usuarios y Usuarias del Mercado de Alimentos (CUUM), especialistas, organizaciones civiles y académicos de varios países latinoamericanos se reunieron para abordar uno de los retos más complejos y prioritarios en salud pública: la influencia de intereses corporativos en las decisiones sobre alimentación y nutrición.
Durante el evento, se destacó la importancia de diseñar políticas públicas libres de conflictos de interés que prioricen la salud de la población por encima de los intereses económicos de la industria alimentaria. Se compartieron las experiencias y estrategias adoptadas en los distintos países para enfrentar este desafío.
Desde Brasil, el Instituto Brasileño de Defensa del Consumidor (IDEC) presentó su trabajo en la protección de los derechos de los consumidores. Esta organización sin fines de lucro, fundada en 1987, ha sido clave en la vigilancia de productos, la promoción de leyes que protegen al consumidor y la lucha por un consumo justo y saludable. Su lema, “Tus derechos, nuestra lucha”, resume su compromiso en este campo.
México estuvo representado por El Poder del Consumidor, una organización que desde 2006 trabaja en campañas informativas, acciones legales y propuestas legislativas para regular la publicidad dirigida a menores, mejorar el etiquetado nutricional y promover una alimentación saludable. Su enfoque integral ha permitido construir alianzas con otras organizaciones e incidir de manera significativa en políticas públicas.
El Instituto Nacional de Salud Pública (INSP) de México también aportó una perspectiva académica. A través de la investigación y la formación de profesionales, el INSP contribuye al diseño de políticas basadas en evidencia científica, especialmente en áreas como nutrición, salud ambiental y enfermedades crónicas. La institución destacó su rol en asesorar al gobierno mexicano y en promover políticas efectivas de salud pública.
Uno de los aportes más relevantes del conversatorio vino de la comunidad NutriCOL, una red de profesionales de la salud comprometida con el trabajo ético y libre de conflictos de interés. NutriCOL hizo énfasis en la necesidad de consolidar una base científica independiente en el desarrollo de políticas públicas, señalando que “la ética en salud pública no es negociable”.
La comunidad destacó su objetivo de impulsar acciones políticas y científicas transparentes para combatir la mala nutrición en todas sus formas. NutriCOL invitó a más profesionales de la salud a sumarse a este compromiso colectivo por sistemas alimentarios justos y sostenibles.
Las conclusiones del conversatorio hicieron un llamado contundente a reforzar las políticas públicas ecuatorianas y regionales frente a los conflictos de interés. Se destacó que el enfoque del gobierno ecuatoriano para abordar este problema de salud pública sugiere que las alianzas público-privadas para la salud son necesarias; sin embargo, primero deben ser transparentes en cuanto a sus estructuras, liderazgo y mecanismos de financiamiento.
También se propuso incluir disposiciones que exijan la rendición de cuentas continua por parte de las organizaciones no gubernamentales (ONG), así como establecer límites claros a la influencia de la industria de alimentos, especialmente a los productores de ultraprocesados y bebidas azucaradas en el diseño y ejecución de políticas nutricionales saludables y que desincentiven su producción y consumo.
El fortalecimiento de mecanismos que favorezcan la transparencia debería exigir la divulgación completa de la composición de todas las entidades—comerciales y no comerciales—que asesoren o participen en la lucha contra la desnutrición infantil y las prácticas alimentarias inadecuadas de la población general.
El conversatorio dejó claro que los conflictos de interés en las políticas alimentarias no son inevitables ni irreversibles: con voluntad política, organización social y evidencia científica, es posible transformarlos en oportunidades para construir sistemas alimentarios más saludables, inclusivos y sostenibles para todos.
Desde Salud y Fármacos consideramos que la protección de la salud pública debe estar por encima de cualquier interés comercial. Las políticas alimentarias deben construirse con base en evidencia científica independiente y propiciar entornos que favorezcan la elección de alimentos saludables, libres de la influencia de actores con intereses económicos, especialmente de la industria de alimentos ultraprocesados y bebidas azucaradas que perpetúan el ciclo sobrepeso-obesidad-prescripción de medicamentos para la pérdida de peso.
Referencias
Crisis en la investigación del Alzheimer: colapso de la hipótesis del β-amiloide y sus implicaciones éticas
Salud y Fármacos
Tags: la hipótesis del β-amiloide en la enfermedad de Alzheimer, fármacos antiamiloides para tratar Alzheimer: Aduhelm (aducanumab), Leqembi (lecanemab) y Kisunla (donanemab), muerte, hemorragias e inflamación cerebral relacionados con fármacos antiamiloide, fraude en la investigación clínica
Durante los últimos 25 años, la investigación sobre la enfermedad de Alzheimer ha sido víctima de una serie de fraudes y otras conductas inapropiadas por parte de investigadores de renombre mundial y de científicos desconocidos [1, 2], todos intentando ascender en un campo brutalmente competitivo.
Entre los escándalos que ha suscitado la I+D en busca de una cura contra la enfermedad de Alzheimer, Piller sigue de cerca lo sucedido con Eliezer Masliah, cuya investigación tuvo una gran influencia en el desarrollo de tratamientos para la pérdida de memoria y la enfermedad de Parkinson, y a quien en 2016 se le confió la dirección del programa del Instituto Nacional sobre el Envejecimiento para abordar el Alzheimer.
El neurocientífico Eliezer Masliah había publicado aproximadamente 800 artículos, muchos de ellos considerados muy influyentes, por lo que parecía ser la persona ideal para dirigir un proyecto para investigar el Alzheimer que contaba con miles de millones de dólares en financiamiento. Sin embargo, en septiembre de 2024, Piller publicó un artículo en la revista Science, describiendo la conducta de Masliah y la evidencia sobre cómo había manipulado fotografías de tejido cerebral y otras imágenes; una clara señal de fraude. Salud y Fármacos resumió el articulo y lo publicó en febrero de 2025 [3].
Pero eso no es todo, Piller solicitó a un equipo de expertos en neuroimagen y en imágenes científicas que lo ayudaran a analizar estudios sospechosos de 46 investigadores destacados en Alzheimer. El proyecto no pretendía realizar un análisis exhaustivo de los 46, y mucho menos de la multitud de otros especialistas en Alzheimer que trabajaron con ellos. Eso requeriría un ejército de detectives y años de trabajo. Pero fue el primer intento de evaluar sistemáticamente el alcance de la manipulación de imágenes por un amplio abanico de científicos clave que investigan una enfermedad.
A lo largo de varios meses, el grupo elaboró un expediente de 300 páginas con 132 artículos del Dr. Masliah que consideraron sospechosos (aunque los artículos fueron escritos en colaboración con colegas, el Dr. Masliah fue el único autor común y, por lo general, desempeñó un papel destacado). Los experimentos incluidos en dichos artículos se habían citado más de 18.000 veces en revistas académicas y médicas.
En conjunto, los expertos identificaron cerca de 600 artículos dudosos que han distorsionado la investigación en este campo; estos artículos se han citado unas 80.000 veces en la literatura científica. Muchos de los estudiosos del Alzheimer más respetados, cuyo trabajo guía el discurso científico, se refirieron repetidamente a estos estudios adulterados para respaldar sus propias ideas.
Piller también ha publicado un artículo en Statnews [4] donde describe como se fue difundiendo la teoría del β-amiloide para explicar la enfermedad de Alzheimer y la búsqueda de tratamientos para dicha enfermedad, y como se fue censurando a los científicos que cuestionaron esa teoría.
Hasta la fecha, ninguna de las hipótesis investigadas explica por sí sola la causa del Alzheimer [5], y solo recientemente se ha empezado a cuestionar la hipótesis del β-amiloide, que ha servido de base para numerosos proyectos de investigación que han costado miles de millones de dólares.
El hecho de que personas con gran cantidad de depósitos de β-amiloide no desarrollen demencia [6] refuerza la importancia de explorar otras hipótesis. Se ha dicho que el papel del β-amiloide podría ser más bien epifenoménico (epifenómeno es un fenómeno accesorio que acompaña al fenómeno principal y que no tiene influencia sobre él [NH1]; es decir, es un subproducto de un proceso, sin un rol causal relevante).
Los medicamentos que se han aprobado recientemente para tratar la enfermedad de Alzheimer eliminan el amiloide con éxito, pero no mejoran significativamente ni la cognición ni ralentizan la velocidad del deterioro, y en algunos casos sus efectos secundarios empeoran la enfermedad de base. Además, cuando hay cambios positivos, son tan pequeños o sutiles que no se consideran clínicamente relevantes y ni los pacientes ni sus cuidadores los perciben.
Si la acumulación de β-amiloide no es la causa del Alzheimer, sino una consecuencia de la enfermedad o una condición relacionada, pero sin un rol activo preponderante en su patogénesis, se desacredita la base científica sobre la que se justifica el desarrollo, aprobación [7] y comercialización de los fármacos antiamiloides para tratar el Alzheimer: Aduhelm (aducanumab), Leqembi (lecanemab) y Kisunla (donanemab).
Esto debería ser suficiente para cuestionar su valor terapéutico, y detener la exposición de adultos mayores a los graves efectos adversos documentados tras el consumo de estos fármacos [8-11]: muerte, hemorragias e inflamación cerebral.
Por otra parte, lo ocurrido nos lleva a resaltar algunos aspectos regulatorios que deberían reformularse para proteger a los pacientes y evitar que se sometan a riesgos innecesarios.
Entre otras cosas se ha documentado la presencia de conflictos de interés [12] entre los miembros de uno de los comités asesores de la FDA y entre el personal de la FDA, así como falta de transparencia y de acceso a los datos de los ensayos clínicos, obstaculizando que grupos independientes y libres de conflictos de interés con la industria puedan verificar los resultados.
También se cuestiona que la FDA otorgara la aprobación acelerada (fast track) en base a una variable indirecta o subrogada, (la reducción de biomarcadores – la presencia de beta-amiloide-), sin evidencia sólida de su correlación con la evolución clínica de la enfermedad.
El detallado reportaje investigativo de Lenzer et al [13] describe múltiples violaciones de los principios éticos fundamentales durante la realización de los ensayos clínicos, como es la falta de equilibrio entre los riesgos y los posibles beneficios [dudosos], la falta de consentimiento verdaderamente informado cuando los participantes aceptaron participar en los ensayos, ya que se omitieron algunos y/o se minimizaron potenciales daños graves.
Adicionalmente, también se ha documentado que cuando han muerto pacientes en tratamiento con antiamiloides, tanto si estaban en ensayos clínicos como en tratamiento para el Alzheimer, no se han hecho autopsias o cuando se han hecho ni siquiera las familias han tenido acceso al resultado, lo cual impide acceder a conocimiento y entorpece el planteamiento de posibles nuevas hipótesis, retrasando el avance científico en esta materia.
Lo sucedido con la búsqueda de tratamientos para el Alzheimer refuerza la necesidad de promover cambios regulatorios, para que esos entes que deben defender el interés público aseguren con vehemencia la proporcionalidad riesgo-beneficio de los tratamientos que autorizan y la publicación íntegra y oportuna de los datos de los ensayos clínicos, incluyendo los de ensayos clínicos negativos. Si expertos independientes hubieran tenido acceso a los resultados de los ensayos clínicos, habría mejorado la posibilidad de explorar otras hipótesis o de diseñar nuevos estudios que evitaran los riesgos y errores ya conocidos.
Desde el punto de vista de política pública, hay que tener en cuenta que estos fármacos antiamiloides, en EE UU [NH2], cuestan alrededor de US$100.000 anuales por paciente (incluyendo infusión, imágenes y seguimiento), por lo que su uso a gran escala podría colapsar los presupuestos de la salud sin generar beneficios proporcionales, lo que tendría un impacto económico insostenible para los sistemas de salud y podría contribuir a que se desviaran fondos que se están utilizando en otras intervenciones más costo-efectivas.
Por último, las extrapolaciones del artículo de Lenzer et al [13] llaman la atención sobre las implicaciones para la salud pública de que se generalice el uso de estos medicamentos por millones de personas con deterioro cognitivo leve o en riesgo de padecerlo, pudiendo resultar en un número significativo de muertes o de personas con secuelas de efectos adversos graves, especialmente ante el crecimiento sostenido de la población envejecida y el aumento mundial de la expectativa de vida.
Referencias