Mar
19
19/03/2026
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Profeco y Cofepris alertan por comercialización ilegal de productos con tirzepatida
Procuraduría General del Consumidor, 11 de marzo de 2026
https://www.gob.mx/profeco/prensa/profeco-y-cofepris-alertan-por-comercializacion-ilegal-de-productos-con-tirzepatida
Tags: falsificación de tirzepatida, comercialización ilegal de tirzepatida, Rapha, Dr. Pen y Peptide Xperts
Se comercializan a través de plataformas de comercio electrónico, sitios web y aplicaciones de dispositivos móviles, sin que cuenten con registro sanitario para su distribución en territorio nacional.
La Procuraduría Federal del Consumidor (Profeco) comparte la alerta que emitió la Comisión Federal para la Protección contra Riesgos Sanitarios (Cofepris) sobre la comercialización ilegal de tres productos con Tirzepatida a través de plataformas de comercio electrónico, sitios web y aplicaciones de dispositivos móviles.
La Cofepris emitió esta alerta sanitaria luego de recibir el análisis técnico-documental de la información presentada por el importador Eli Lilly y Compañía de México, S.A. de C.V., que identificó la venta ilegal.
Se trata de los productos marca Rapha, Dr. Pen y Peptide Xperts que se ofertan en distintas presentaciones, como gotas, parches, viales, jeringas prellenadas, sin que cuenten con registro sanitario para su distribución en territorio nacional.
La Cofepris subraya que la adquisición y uso de tirzepatida debe realizarse bajo prescripción, de conformidad con el artículo 226 de la Ley General de Salud que clasifica los medicamentos para su venta y suministro y advierte que su uso indiscriminado y sin supervisión de un médico especialista, puede ocasionar un estrés o sobreactividad en órganos como el hígado o el páncreas, entre otros, lo que puede generar un fallo en los mismos.
Ante el riesgo, la institución recomienda nunca adquirir estos productos en cualquier presentación y en caso de contar con información sobre su posible comercialización, realizar la denuncia sanitaria correspondiente.
En caso de estar utilizando los productos mencionados, se recomienda suspender de inmediato su uso y consultar con un profesional de la salud para una valoración médica.
Si alguna persona ya lo ha consumido y presenta reacciones adversas, se recomienda que lo reporte en el siguiente enlace: https://vigiflow-eforms.who-umc.org/mx/vigiramo al correo electrónico:farmacovigilancia@cofepris.gob.mx.
La alerta puede consultarse en: https://www.gob.mx/cms/uploads/attachment/file/1055652/Alerta_Sanitaria_Tirzepatida_09022026.pdf.
Alerta Sanitaria COFEPRIS. Comercialización ilegal del producto trikafta.
(elexacaftor, tezacaftor e ivacaftor), 100 mg, 50 mg y 75 mg; (ivacaftor) 150 mg comprimidos.
Comisión Federal para la Protección contra Riesgos Sanitarios, 3 de marzo de 2026
https://www.gob.mx/cms/uploads/attachment/file/1060524/Alerta_Sanitaria_trikafta_03032026.pdf
Tags: Trikafta, elexacaftor, tezacaftor, ivacaftor, comercialización ilegal de trikafta
Categoría: Alerta sanitaria de medicamentos
Lugar de expedición: Ciudad de México
La Comisión Federal para la Protección contra Riesgos Sanitarios (COFEPRIS) informa sobre la comercialización ilegal del producto trikafta® (elexacaftor, tezacaftor e ivacaftor) 100 mg, 50 y 75 mg; (ivacaftor) 150 mg, comprimidos.
Esta alerta sanitaria se emite derivada del análisis técnico-documental con base en la información proporcionada por el representante legal en México, que identificó la comercialización ilegal del producto trikafta® con número de lote W077103 y fecha de caducidad 04-2026, destinado originalmente a Uruguay.
Sin embargo, al comercializarse en territorio nacional, se desconoce si el producto fue importado ilegalmente o podría tratarse de una falsificación, debido a su bajo costo. Cabe destacar que, al ser un producto destinado a otro país, no cumple con el etiquetado autorizado para su venta en México, por lo que se desconocen las condiciones en las que fue importado, manipulado, almacenado y distribuido, lo que representa un riesgo para la salud de la población, ya que no se garantiza su calidad, seguridad y eficacia.
Por lo anterior, COFEPRIS emite las siguientes recomendaciones:
Población en general y profesionales de la salud:
Distribuidores y farmacias:
COFEPRIS mantendrá acciones de control sanitario e informará oportunamente a la población en caso de identificar nuevas evidencias, con el fin de prevenir riesgos a la salud de la población, procedentes de productos, servicios o establecimientos que incumplan con la legislación sanitaria vigente.
“El presente, se emite con fundamento en los artículos 4°, párrafo cuarto, de la Constitución Política de los Estados Unidos Mexicanos; 17 y 39, fracción XXI, de la Ley Orgánica de la Administración Pública Federal; 1, 17 Bis, fracción I, de la Ley General de Salud; y 3, 12 del Reglamento de la Comisión Federal para la Protección contra Riesgos Sanitarios.”
“De conformidad con el derecho a la protección a la salud y por principio precautorio, la Comisión Federal para la Protección contra Riesgos Sanitarios con la finalidad de reducir los riesgos sanitarios a la población, informa sobre situaciones que presentan un riesgo sanitario para la salud de la población en general, derivado del uso y/o consumo de diversos insumos para la salud, productos o servicios que no cumplen con la regulación sanitaria vigente.”
Arixtra: La AEMPS informa sobre un defecto de calidad y las precauciones a adoptar
Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), 6 de febrero de 2026
https://www.aemps.gob.es/informa/la-aemps-informa-sobre-un-defecto-de-calidad-en-el-medicamento-arixtra-y-las-precauciones-a-adoptar/
Tags: Viatris Pharmaceuticals, Arixtra, defecto de calidad Arixtra, eventos tromboembólicos venosos
Categoría: Medicamentos de uso humano, defectos de calidad
Referencia: ICM (CONT), 04/2026
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha tenido conocimiento, a través del titular de autorización de comercialización del medicamento Arixtra, de un defecto de calidad en la aguja de las jeringas precargadas de este medicamento (ver foto).
El defecto ha sido detectado tras recibir notificaciones relacionadas con una coloración marrón y obstrucción en la aguja de las jeringas precargadas de este medicamento indicado para prevenir que se formen coágulos de sangre en los vasos sanguíneos. La causa de este defecto está relacionada con la presencia de una partícula de hierro que se ha oxidado en el interior de la aguja.
Actualmente, la investigación está en curso para identificar la causa raíz e implementar las acciones correctivas y preventivas correspondientes.
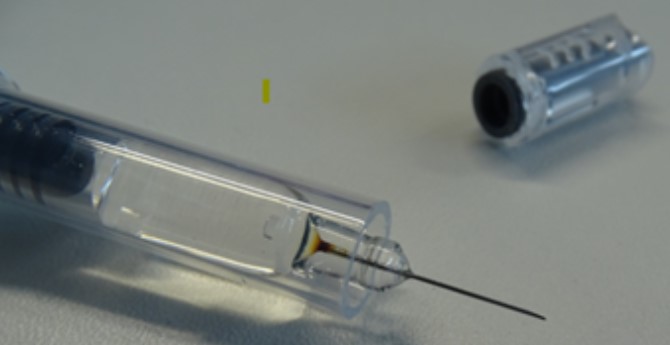
Se estima que el defecto es muy poco frecuente, no habiéndose recibido ninguna reclamación relacionada con este evento en España, y podría aparecer en cualquier lote de las presentaciones de Arixtra comercializadas en nuestro mercado:
Los riesgos potenciales del uso de una jeringa precargada con presencia de óxido incluyen falta de eficacia por obstrucción de la aguja, reacciones adversas si se administra la inyección afectada (reacciones de hipersensibilidad, complicaciones en el lugar de inyección, incluida rotura de aguja, efectos tromboembólicos e infecciones sistémicas).
Dependiendo de la dosis, Arixtra está indicado para:
Información para profesionales sanitarios
Información para farmacias
Datos de la empresa
Viatris Pharmaceuticals, S.L.
(+34) 900 102 712
Correo electrónico para notificar defectos de calidad: cliente@viatris.com, ReclamacionesQA.Spain@viatris.com
Correo electrónico para notificar sospechas de reacciones adversas: phvg.spain@viatris.com
Reino Unido. La MHRA actualiza la guía de GLP-1 para prescriptores y pacientes
(MHRA updates guidance for GLP-1 prescribers and patients)
Agencia Reguladora de Medicamentos y Productos Sanitarios (MHRA), 29 de enero de 2026
https://www.gov.uk/government/news/mhra-updates-guidance-for-glp-1-prescribers-and-patients
Traducido por Salud y Fármacos
Tags: actualización guía GLP-1, pancreatitis aguda, Wegovy, Ozempic, Mounjaro, semaglutida, tirzepatida
Actualización de la información de los fármacos GLP-1 para los profesionales de la salud y pacientes para informar sobre el bajo riesgo de pancreatitis aguda grave asociado a consumo de fármacos GLP-1.
La Agencia Reguladora de Medicamentos y Productos Sanitarios (MHRA por su sigla en inglés), actualizó hoy (29 de enero de 2026) la información del producto dirigida a profesionales de la salud y a pacientes, para informar sobre el bajo riesgo de pancreatitis aguda grave en personas que reciben tratamiento con agonistas del receptor del péptido similar al glucagón tipo 1 (GLP-1) o con agonistas duales del receptor GLP-1/polipéptido insulinotrópico dependiente de la glucosa (GIP), comúnmente conocidos como GLP-1.
La pancreatitis aguda constituye una reacción adversa conocida, aunque poco frecuente, asociada al tratamiento con GLP-1. En casos extremadamente raros, algunos pacientes pueden presentar complicaciones de una pancreatitis aguda muy graves. Se recuerda a médicos y pacientes la importancia de permanecer atentos a los síntomas iniciales, entre ellos, el dolor abdominal intenso y persistente, que puede irradiarse a la espalda y acompañarse de náuseas y vómitos.
Los GLP-1 se prescriben para el tratamiento de la diabetes tipo 2 y, en el caso de determinados productos, para el control del peso corporal y la reducción del riesgo cardiovascular en personas con enfermedad cardiovascular establecida y un índice de masa corporal (IMC) igual o superior a 27 kg/m².
Una investigación publicada recientemente por el University College de Londres estima que 1,6 millones de adultos en Inglaterra, Gales y Escocia utilizaron GLP-1, como semaglutida (Wegovy, Ozempic) y tirzepatida (Mounjaro), entre principios de 2024 y principios de 2025 para bajar de peso [1].
Si bien los GLP-1 se consideran, en general, medicamentos seguros y eficaces dentro de sus indicaciones autorizadas, como ocurre con todos los medicamentos, no están exentos de riesgos. Las personas que reciben tratamiento con GLP-1 deben conocer los síntomas de pancreatitis grave y buscar atención médica urgente ante la aparición de esos síntomas.
Alison Cave, Directora de Seguridad de la MHRA, afirmó:
“La seguridad del paciente es la máxima prioridad de la MHRA y monitoreamos de forma continua la seguridad y eficacia de todos los medicamentos autorizados. Para la gran mayoría de los pacientes a los que se les prescriben GLP-1, estos son medicamentos seguros y eficaces que ofrecen beneficios relevantes para la salud. El riesgo de desarrollar estos efectos secundarios graves es muy bajo, pero es importante que los pacientes y los profesionales de la salud estén atentos y reconozcan los síntomas asociados”.
“Si usted o un ser querido está tomando GLP-1 y nota síntomas como un dolor de estómago intenso y persistente que puede irradiarse a la espalda y/o estar acompañado de náuseas y vómitos, le recomendamos que consulte con un profesional de la salud y lo notifique a través de nuestro programa Yellow Card [2].”
El Biobanco Yellow Card, una iniciativa conjunta de la MHRA y Genomics England, ha reclutado pacientes en tratamiento con GLP-1 con el objetivo de investigar si el riesgo de complicaciones graves por la inflamación aguda del páncreas podría verse influido por factores genéticos individuales [3, 4].
Se espera que esta investigación contribuya a identificar a los pacientes con mayor riesgo de reacciones adversas y a apoyar la prescripción de tratamientos más seguros y personalizados.
La guía sobre los posibles efectos secundarios de los GLP-1 y su uso seguro y eficaz está disponible en el sitio web de la MHRA.
Referencias:
ALERTA DIGEMID Nº 011- 2026: Retiro del mercado del lote Nº 2304067 del producto MEGACILINA B 1.200.000 UI Polvo para suspensión inyectable (Bencilpenicilina benzatínica), por resultado crítico de control de calidad. Caja con un vial con polvo para reconstituir con 1 ampolla con disolvente (agua estéril) por 5 mL Registro Sanitario N° EE-05782, Lote disolvente 2302564
Dirección General de Medicamentos, Insumos y Drogas (DIGEMID), 27 de enero de 2026
https://www.digemid.minsa.gob.pe/Archivos/Alertas/2026/ALERTA_11-26.pdf
Tags: alerta Megacilina B, fallos de calidad, penicilina benzatínica, bencilpenicilina
La Dirección General de Medicamentos, Insumos y Drogas (Digemid) del Ministerio de Salud, comunica a los profesionales de la salud, instituciones, establecimientos farmacéuticos, responsables del suministro de medicamentos en el sector público y privado, y al público en general lo siguiente:
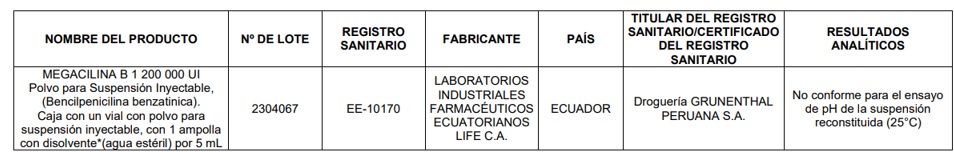
Existiendo la posibilidad de que algún producto del lote observado permanezca en posesión del usuario final, la Digemid realiza la presente comunicación a fin de evitar su uso y salvaguardar la salud de la población.
Para más información sobre resultados de control de calidad verificar en el siguiente enlace: http://observatorio.digemid.minsa.gob.pe/obscalidad/webform2
El organismo regulador indio rechaza la exención de los ensayos clínicos de fase III locales con lenacapavir
(Indian regulator declines local Phase-III clinical trial waiver for Lenacapavir).
Chetali Rao, K.M. Gopakumar
Third World Network, 13 de noviembre de 2025
www.twn.my
Traducido por Salud y Fármacos, publicado en Boletín Fármacos: Propiedad Intelectual 2026; 29 (1)
Tags: atrasos en el acceso a lenacapavir, acceso mundial a versiones genéricas de lenacapavir, ensayos clínicos con lenacapavir
El comité de expertos de la agencia reguladora de medicamentos de India ha rechazado la solicitud de Hetero Lab Ltd. Hetero aspiraba a que la eximiera de realizar dos estudios y le permitiera fabricar y comercializar lenacapavir genérico para la prevención y el tratamiento del VIH [1].
El Comité de Expertos en la Materia (SEC) de la Organización Central de Control de Estándares de Medicamentos (en inglés Central Drugs Standard Control Organization CDSCO) rechazó la solicitud de Hetero y no le otorgó el permiso para fabricar ni comercializar el comprimido de lenacapavir de acción prolongada de 300 mg. Las actas de la reunión [1] señalan que la propuesta de Hetero justificaba la exención de hacer un ensayo clínico de fase III (local) y de un estudio de bioequivalencia (BE).
Hetero se encuentra entre los seis fabricantes de genéricos que han recibido una licencia voluntaria, libre de regalías, de Gilead Sciences para fabricar y vender lenacapavir genérico [2].
La Regla 75 de las Normas sobre Nuevos Medicamentos y Ensayos Clínicos (2019) [3] incorpora una disposición importante para la exención de ensayos clínicos locales en India. La Regla 75 establece que:
“No será necesario presentar el ensayo clínico local junto con la solicitud a la que se refiere el apartado (1) si:
Si bien Hetero solicitó una exención en base al gran número de ensayos clínicos con lenacapavir realizados a nivel mundial, el comité observó que Hetero no presentó datos de estudios clínicos sobre la subpoblación india que demostraran que no hay una variabilidad étnica sustancial. El comité consideró que la evidencia era insuficiente para confirmar la adecuación de la dosis y su seguridad/eficacia ni aprobar la dosis propuesta para la población india. Esto significa que Hetero tendrá que realizar un estudio clínico local de fase III en la población india antes de obtener la aprobación para lenacapavir. Sin embargo, la empresa ya había obtenido permiso para la realización de un estudio de bioequivalencia (BE) con fines de exportación de la división de bioequivalencia/biodisponibilidad de la CDSCO. Dicho estudio de bioequivalencia está en curso.
El rechazo parece discrepar de las Normas de Ensayos Clínicos con Medicamentos Nuevos de 2019
Como se informó anteriormente [4], lenacapavir no se ha registrado en India y no aparece en la lista de nuevos medicamentos aprobados en India en 2024 [5] y 2025 [6]. Una orden de la Autoridad Central de Licencias (CLA) con fecha 7 de agosto de 2024 [7], emitida en virtud de la Regla 101 de las Normas de Medicamentos Nuevos y Ensayos Clínicos, especifica que si un nuevo medicamento está aprobado y comercializado en EE UU, Reino Unido, UE, Japón, Australia o Canadá, se puede considerar la exención de ensayos clínicos locales para las siguientes cinco categorías: medicamentos huérfanos, terapias génicas/celulares, medicamentos utilizados en situaciones de pandemia, medicamentos para fines especiales de defensa y medicamentos que ofrecen un avance terapéutico significativo con respecto al estándar de atención actual.
Lenacapavir cumple los requisitos para ser considerado en virtud de la Regla 101 por las siguientes razones:
Existen precedentes [8] en los que la SEC, con la posterior aprobación del Comité Técnico/Comité Superior, ha recomendado exenciones en circunstancias excepcionales cuando existían datos suficientes de ensayos de Fase III a nivel mundial y se impusieron condiciones de farmacovigilancia posterior a la comercialización. Esto incluye medicamentos para el VIH y la tuberculosis. En 2015 también se concedió una exención de ensayos clínicos locales para un fármaco antiviral contra el VIH.
En los ensayos clínicos pivotales, incluyendo los estudios PURPOSE 1 y PURPOSE 2 [9], el lenacapavir demostró ofrecer una protección casi completa contra la adquisición del VIH, con tasas de eficacia del 96-100 % y un sólido perfil de seguridad. Existen sólidos datos globales de Fase III para lenacapavir (tanto para el tratamiento como para la prevención) en múltiples países y poblaciones [10].
Si bien el estudio PURPOSE 1 se realizó en Sudáfrica y Uganda, PURPOSE 2 se llevó a cabo en EE UU, Sudáfrica, Perú, Brasil, Argentina, México y Tailandia. Si la población extranjera es similar a la población india en parámetros clave (como el subtipo de VIH y las comorbilidades), la necesidad de ensayos clínicos se reduciría. En cuanto al subtipo de VIH, un estudio ha revelado que, en el sur de África, Etiopía y el sur de Asia (India entre otros países) predominaba el subtipo C, que constituía al menos el 97 % de las infecciones en cada región entre 2016 y 2021 [11]. Además, dados los perfiles de comorbilidad, como la alta tasa de coinfección por tuberculosis en ambas poblaciones, los datos internacionales respaldaron la exención en India. Además, la etiqueta de la FDA no reporta diferencias clínicamente significativas en la farmacocinética de lenacapavir según la edad, el sexo, la etnia, la raza o el peso corporal, lo que reduce la necesidad de hacer un estudio en la población india [12]. La población del ensayo global ya era diversa, incluyendo cohortes latinoamericanas y africanas, lo que habría cubierto las posibles variaciones.
Dada la carga de VIH en la India (más de 60.000 infecciones anuales) y el objetivo de contar con herramientas de prevención de acceso rápido, los argumentos de salud pública favorecen una aprobación acelerada de lenacapavir. Lenacapavir representa un avance transformador en la prevención y el tratamiento del VIH, ofreciendo la primera y única profilaxis preexposición (PrEP) inyectable semestral, un medicamento para prevenir el VIH. Para las personas que viven con el VIH, el lenacapavir representa una opción crucial, especialmente para aquellas con virus multirresistente. Su formulación de acción prolongada supera una de las barreras más importantes en la atención del VIH: la adherencia subóptima a los medicamentos diarios, particularmente entre las poblaciones afectadas por el estigma, la inestabilidad social o el acceso limitado a la atención médica.
Sin la exención, el plazo para la introducción de lenacapavir genérico en la India se ampliará. Los retrasos en el acceso no solo afectarán a la India, sino también a una gran cohorte de países de ingresos bajos y medianos (PIBM) cubiertos por la licencia de Gilead, muchos de los cuales dependen de los genéricos indios para su suministro. Otorgar esta exención no solo habría acelerado el acceso a lenacapavir para los 2,3 millones de personas que viven con VIH en India, sino que también habría posicionado a India como proveedor global, evitando retrasos en la implementación en los PIBM. Este obstáculo regulatorio pospone los posibles beneficios para la salud pública que podría ofrecer un lenacapavir genérico oportuno y asequible.
Referencias
Más allá de la resistencia: modelos alternativos de innovación para el acceso global y la gestión de nuevos antibióticos
(Beyond resistance: alternative innovation models for global access and stewardship of new antibiotics)
Slovenski, I., Alonso Ruiz, A., Vieira, M. et al.
Humanit Soc Sci Commun 2025;12, 2006. https://doi.org/10.1057/s41599-025-06337-y
https://www.nature.com/articles/s41599-025-06337-y libre acceso en inglés
Traducido por Salud y Fármacos
Tags: acceso equitativo a los antibióticos, gestión de nuevos antibióticos
Resumen
Para proteger la salud pública y abordar la resistencia a los antimicrobianos (RAMI) es muy importante acelerar el desarrollo, promover el acceso equitativo y gestionar adecuadamente los antibióticos nuevos.
Para entender y evaluar cómo el actual subsistema de innovación en antibióticos, o “nicho”, aborda esta tarea, revisamos la literatura, compilamos y analizamos una base de datos de 211 desarrolladores de antibióticos y realizamos 10 entrevistas con las partes interesadas.
Descubrimos que una minoría de desarrolladores de antibióticos sigue utilizando l modelo convencional de innovación impulsado por el mercado, pero la mayoría adopta uno de los dos modelos alternativos:
Este modelo está compuesto por actores públicos y privados, respaldados por organizaciones filantrópicas y/o sin fines de lucro como Wellcome, CARB-X (por su sigla en inglés Combating Antibiotic-Resistant Bacteria Biopharmaceutical Accelerator) o GARDP (por su sigla en inglés Global Antibiotic Research and Development Partnership).
Concluimos que el modelo de innovación alternativo de RC representa el desarrollo más prometedor para garantizar la innovación con acceso y gestión responsable. Sin embargo, el modelo es relativamente reciente y aún debe demostrar su capacidad para lograr estos resultados.
Las reglas y normas que rigen el nicho de los antibióticos están evolucionando y las decisiones políticas, como la adopción de incentivos de atracción (pull incentives), pueden influir profundamente en qué modelos podrían tener éxito en el futuro.
Los responsables de tomar decisiones deben elaborar reglas e incentivos que establezcan las condiciones propicias para que los modelos conjuntamente aporten innovación, acceso y gestión responsable de los antibióticos, para ayudarlos a mejorar, sobrevivir y prosperar.
Retiro del mercado estadounidense de Ixchiq, vacuna contra el virus del chikungunya
Salud y Fármacos
Tags: seguridad de Ixchiq, vacuna viva contra chikungunya, licencia de Ixchiq, suspensión de Ixchiq
Tras la decisión de la FDA de intensificar la evaluación de seguridad de la vacuna contra el chikungunya, la empresa farmacéutica Valneva retiró del mercado estadounidense su vacuna Ixchiq.
La medida siguió a la suspensión del estudio post comercialización y a la previa revocatoria de la licencia del biológico, motivadas por la identificación de eventos adversos graves, incluyendo más de veinte casos de enfermedad tipo chikungunya y un fallecimiento por encefalitis atribuible a la vacuna.
El Centro de Evaluación e Investigación de Productos Biológicos (CBER) concluyó que, en la mayoría de los escenarios, los riesgos del biológico superaban sus beneficios, por lo que su uso continuado podría representar un riesgo para la salud pública.
Aunque Ixchiq había mostrado eficacia en los ensayos clínicos y estaba indicada para adultos con riesgo de exposición al virus en zonas endémicas, la FDA mantuvo su decisión regulatoria. La FDA también ha aprobado una vacuna alternativa basada en partículas similares al virus, recomendada para viajeros a áreas con brotes.
Nota de SyF: Amplíe aquí la información sobre laseguridad de Ixchiq y la suspensión de la licencia por parte de la FDA: https://www.saludyfarmacos.org/boletin-farmacos/boletines/nov202501/06_ac/
Fuente Original:
Residuos de medicamentos en el agua: la industria farmacéutica regatea su eliminación
(Drug residues in water: the pharmaceutical industry haggling over their removal)
Prescrire International 2025; 34 (275): 255
Traducido por Salud y Fármacos
Tags: contaminación del agua, Tratamiento de las Aguas Residuales Urbanas, equilibrio de ecosistemas acuáticos, Pacto Verde, obligaciones de la industria farmacéutica
Entre las diversas directivas europeas sobre la contaminación del agua, la revisión de la Directiva sobre el Tratamiento de las Aguas Residuales Urbanas entró en vigor a principios de 2025 [1]. La industria farmacéutica está intentando evadir sus obligaciones.
La Directiva 2024/3019 del 27 de noviembre de 2024 establece muchas obligaciones para las autoridades locales y ciertas industrias. Se han fijado una serie de fechas objetivo, entre 2025 y 2045, para que las plantas de tratamiento implementen distintos niveles de tratamiento de las aguas residuales urbanas: tratamiento primario, que reduce la cantidad de sólidos en suspensión; tratamiento secundario, que reduce la materia orgánica; tratamiento terciario, que reduce la cantidad de fósforo y nitrógeno (que alteran el equilibrio de los ecosistemas acuáticos); y el tratamiento cuaternario, que reduce la cantidad de micro contaminantes [1, 2].
La Comisión Europea consideró que los medicamentos representan el 59% de los micro contaminantes encontrados en las aguas residuales (a través de su excreción en orina y heces), y que los productos cosméticos representan el 14% [3]. Siguiendo el principio de “el que contamina, paga”, la directiva posibilita que estos dos sectores industriales financien el 80% de las actividades destinadas a eliminar los micro contaminantes del agua —cuyo coste total se estimó en €1.200 millones al año—, y que el 20% restante sea financiado por el sector público o con tasas por el agua [1, 3].
Las plantas de tratamiento necesarias para reducir la cantidad de micro contaminantes son principalmente las que tratan las aguas residuales de una población que supere los 150.000 habitantes: deben estar operativas para 2045 [1]. Menos del 1% de las plantas de tratamiento de aguas residuales que hay en Francia entran en esta categoría [4].
En marzo de 2025, representantes de la industria farmacéutica anunciaron su intención de impugnar la directiva ante el Tribunal General de la Unión Europea [5, 6]. La industria considera que, al dirigirse solo a dos sectores industriales, la directiva va en contra de los principios europeos de “el que contamina, paga”, de proporcionalidad y de no discriminación, y que la Comisión ha subestimado considerablemente el coste que la industria farmacéutica tendría que pagar por el tratamiento [6].
Quizá la industria farmacéutica vio una oportunidad, ya que recientemente la Comisión Europea ha tendido a favorecer a las empresas europeas: a principios de 2025, suavizó las obligaciones que el Pacto Verde impone a las empresas [7]. El futuro de la directiva está ahora en manos de los tribunales europeos.
Mientras tanto, si hubiera que reescribir la directiva, las plantas de tratamiento de aguas residuales seguirán liberando residuos de medicamentos en el medioambiente.
Referencias

