Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
BIOEQUIVALENCIA, AMBIGÜEDADES, OPORTUNISMO Y EL CASO DEL RETIRO DE ARV DE LA LISTA DE PRECALIFICACIÓN DE LA OMS (a)
Claudia Vacca1 y Francisco Rossi2
1 Programa de Formación en Salud Internacional, Washington, DC,
2 Internacional Poverty Centre, UNDP
El caos y el oportunismo alrededor de la decisión del retiro de 5 medicamentos antirretrovirales de la lista de precalificación de la OMS, entre junio y agosto de este año, eran acontecimientos tan previsibles como evitables, dada la continua y creciente desinformación que rodea el tema de la bioequivalencia y representan a la vez, la mejor fotografía de las tensiones y las sinergias que se entretejen en las discusiones de calidad y acceso.
Desde que la OMS adoptó los criterios que debían cumplir los productos que requerían pruebas de bioequivalencia [1], los países han realizado libre interpretación, adaptación u omisión de los mismos, concientes de que los determinantes de la calidad farmacéutica se centran en la certificación de las Buenas Prácticas de Fabricación (BPF).
En efecto, un estudio realizado en 6 países de la región de las Américas sobre los productos a los que se les exige estudios de bioequivalencia, realizado dentro de la Red Panamericana de Armonización Farmacéutica (RED PARF cuya secretaría técnica está en cabeza de la Organización Panamericana de la Salud, OPS), estableció que solo 4 productos son comunes entre los países estudiados [2]. En un análisis realizado por la misma Red, cuantificando el riesgo sanitario de medicamentos comercializados para establecer la necesidad de los estudios de bioequivalencia, el percentil 20 de la lista, contiene solo 15 productos, dentro de los cuales no se encuentran los antirretrovirales [3].
Mientras tanto, los representantes de intereses comerciales, hacen su propia lectura. Para las multinacionales el requisito fundamental para que un producto competidor pueda ser considerado de categoría similar al innovador y por lo tanto pueda ser sustituido, es la prueba de bioequivalencia, no importa si el producto representa o no un riesgo sanitario.
Aunque el requisito de bioequivalencia opera en el ámbito nacional, como parte de los procesos de registro, su exigencia ha traspasado ámbitos regionales. Por ejemplo, en la negociación de precios de Antirretrovirales del Área Andina, los negociadores nacionales tuvieron que incorporar el requisito de bioequivalencia, aunque ello representara la eliminación de proveedores de genéricos producidos en Latinoamérica y precios significativamente menores [4].
Precisamente, la decisión de retiro de antirretrovirales de la lista de precalificación de la OMS, producidos por dos diferentes laboratorios de la India, se debió a la incertidumbre sobre la bioequivalencia de los productos [5].
El comunicado de la OMS establece que, aunque no se sabe si los productos son bioequivalentes, estos son de calidad dado que respondieron a los estándares de especificaciones del principio activo, perfil de impurezas, formulación y fabricación en cumplimiento de las (BPF).
En otras palabras, la salida de los productos se debe a un requisito que no es determinante de la calidad. De manera que, es pertinente preguntarse el papel de las pruebas de bioequivalencia en la evaluación de la calidad farmacéutica y la razón de su incorporación al proceso de precalificación.
Este documento presenta los antecedentes de la decisión del retiro de algunos antirretrovirales de la lista de precalificación de la OMS y de los debates sobre el papel de las pruebas de bioequivalencia. También espera convertirse en una invitación a profundizar los debates éticos, técnicos y políticos asociados a la exigencia de la bioequivalencia como requisito de calidad de los productos genéricos.
El retiro de 5 antirretrovirales de la lista de precalificación de la OMS: antecedentes
El proyecto de precalificación de medicamentos prioritarios, es el mecanismo del Sistema de Naciones Unidas (UNICEF, el Banco Mundial, ONUSIDA y otras agencias) de compra de medicamentos para los programas de SIDA, Malaria y Tuberculosis. La precalificación da origen a una lista de productos y proveedores que cumplen con las normas y estándares establecidos en las guías de la OMS (complementadas por la Internacional Conference of Harmonization -ICH- cuando corresponda) [6].
La lista de productos precalificados, que empezó siendo una guía para los países que contaban con autoridades reguladoras débiles, terminó convirtiéndose en el referente obligatorio de calidad para los beneficiarios de dineros del Fondo Global, a pesar de la existencia de regulaciones nacionales en materia de registro y de capacidades nacionales en materia de registro, vigilancia y control de calidad.
Cualquier sistema de precalificación debe estar diseñado para “filtrar o capturar” productos de dudosa o mala calidad y, puede constituirse en el principal elemento de confianza para los usuarios y la comunidad. Sin embargo, lo que pasó con el retiro de los productos, en palabras de la OMS, no fue que los productos incumplieran con los criterios de calidad.
Esta incertidumbre se presentó por que el proceso de precalificación incorpora la presentación de las pruebas de bioequivalencia y dichas pruebas fueron contratadas a una institución externa por el laboratorio productor. La institución no cumplió con algunos requisitos de las buenas prácticas clínicas y de laboratorio en la inspección de la OMS, con lo cual ésta no puede afirmar si el producto es bioequivalente o no [7].
Por supuesto, en coherencia con un sistema de evaluación, aún si una mínima parte de un proceso de inspecciones y verificaciones falla, deben tomarse medidas al respecto. Pero las medidas deben tomarse en relación con el efecto y las consecuencias.
La reacción ante la comunicación de la OMS no ha hecho esperar. Las organizaciones de personas viviendo con VIH/SIDA, presionadas con información sistemática sobre la dudosa calidad de los medicamentos genéricos, exigieron las pruebas de bioequivalencia para los medicamentos [8].
Los laboratorios transnacionales, con nuevos ánimos y energías y con una copia de la decisión de la OMS, han iniciado una campaña institucional, individualizada y de lobby en contra de los antirretrovirales genéricos.
Los países beneficiarios del fondo global, obligados a comprar productos precalificados, se encontraron ante una situación compleja y riesgosa. Por un lado el fondo global y los pacientes exigiendo productos precalificados bioequivalentes, por otro intentando consumir las cantidades de productos abastecidas antes de la decisión y finalmente, evitando los riesgos derivados de posibles interrupciones del tratamiento.
Situación compleja que contiene respuestas simples: si existen países, en donde se comercializan y usan medicamentos antirretrovirales sin pruebas de bioequivalencia, hay resultados favorables en el aumento de la supervivencia, la disminución de las tasas de mortalidad y bajas tasas de resistencia; los pacientes podrían aceptar y continuar su tratamiento sin interrupciones [9].
El asunto es que, como dice la comunicación de una ONG internacional, la OMS pidió a los fabricantes la presentación de otros estudios y si estos cumplen con las exigencias, los productos deberían ser reincorporados en la lista de precalificación. Por lo tanto se pensaría que la OMS tomó una decisión apresurada, con graves consecuencias [10].
Además, en una muestra de eclecticismo confuso, la OMS/OPS incluyó la recomendación de la no interrupción de la terapia, mientras los países se reabastecían con otros productos precalificados, con lo cual el núcleo del debate persiste en la necesidad o no de las pruebas de bioequivalencia.
Ambigüedades y peligros de los criterios contenidos en el informe 34 de la OMS
Los criterios del informe de la OMS sobre el tema de la bioequivalencia, conocido como informe 34, son generales en su contenido y a la vez ambiguos. Por ejemplo, sobre la definición de medicamento genérico, reconoce la diversidad de los países y la relación de la definición con la evolución de sus propias regulaciones, por lo tanto prefiere usar el término producto farmacéutico de fuentes múltiples [11].
Sin embargo, establece que el uso del término “producto genérico” hace referencia a un producto farmacéutico, generalmente intercambiable con el producto innovador, que por lo común se fabrica sin licencia de la empresa innovadora y se comercializa tras haber vencido la patente u otros derechos de exclusividad [12]. Con lo cual adopta una definición de medicamento genérico que refleja la realidad de aquellos países en los que los genéricos surgieron en el marco de un sistema de patentes operante y en donde las pruebas de bioequivalencia son un requisito extendido de comercialización.
Pero esa no es la única, ni más grave ambigüedad. La más grave la representa el hecho de establecer una serie de criterios que justifican la realización de la prueba de bioequivalencia para ciertos productos. Es decir, acepta que no todos los medicamentos requieren demostrar bioequivalencia, más allá de las excepciones obvias (soluciones intravenosas o formas líquidas orales).
Sin embargo, establece que el medicamento genérico que espere ser intercambiable deberá realizar un estudio de bioequivalencia. En otras palabras, hay algunas razones de importancia clínica, por la que se requeriría un estudio que demostrara bioequivalencia entre un producto de referencia y un producto competidor. Sin embargo, aún cuando no se requiera, debería realizarse solo para que el producto competidor adquiera el derecho de reemplazar el producto de referencia.
Esta última ambigüedad es el asunto de fondo de la manipulación de una información, que con pretensiones técnicas, se convierte hoy en una de las más peligrosas barreras de acceso a los medicamentos, particularmente a los antirretrovirales.
Paradojas asociadas a la calidad
La paradoja consiste en que algunos que defienden el acceso a los medicamentos, mediante la promoción de la competencia, también lo hacen exigiendo que los competidores hayan realizado la prueba de bioequivalencia, confundiendo esta exigencia con la necesaria demostración de calidad.
Calidad no es bioequivalencia y bioequivalencia no es calidad. La calidad es una condición de los productos farmacéuticos según la cual existen unos requisitos básicos obligatorios por debajo de los cuales el riesgo de comercializar un producto es inaceptable.
Hay un consenso internacional en que el conjunto básico de requisitos están contenidos en la certificación de las BPF, que incorpora la evaluación y verificación de las materias primas y las pruebas de disolución (en el caso de los sólidos orales).
Para los medicamentos, los criterios básicos obligatorios de registro, no distinguen en la fuente de producción y eliminan el concepto errado de medicamentos para pobres y medicamentos para ricos, genéricos e innovadores, innovadores y competidores o las odiosas diferencias entre similares, copias e innovadores.
Cualquiera de las distinciones olvida que todos los medicamentos son, en su comienzo y substancia, “genericos”, es decir principios activos, a los cuales se añade, por razones de mercado, el nombre de marca y que, clausurada la puerta del mercado, recuperan sus características básicas. El debate sobre sobre la bioequivalencia, en términos de intercambiabilidad, no es un problema médico, si no de mercado, una relación dinámica entre producción, distribución, dispensación y costos [13].
Diferentes acercamientos a la bioequivalencia.
El estudio de bioequivalencia, es un método clínico, que se realiza sobre un número reducido de voluntarios sanos y mide comparativamente la cantidad de sustancia activa de dos productos, uno de referencia y otro de comparación, que llega a la sangre. La medición comparativa permite presumir que, si la diferencia no es estadísticamente significativa, los productos tendrán iguales efectos [14]. O sea que, el estudio de bioequivalencia es una prueba intermedia, predictiva, del desempeño terapéutico.
Algunos creen que éticamente este estudio es cuestionable porque no cumple el principio de beneficencia (no espera demostrar que un producto es superior a otro sino simplemente igual).
Otros creen que solo debe realizarse, si es indispensable clínicamente, es decir relevante en términos de salud pública. En este sentido, un estudio de bioequivalencia es una tasa mínima (un sobrecosto) que se paga, por arte de estructuras de mercado, como un requisito de registro, solo en los casos en los cuales es relevante y no debe ser trasformados en objeto de confrontaciones falsamente “técnicas” [15]. Debe anotarse que los medicamentos genéricos de India y Canadá encuentran en el mercado norteamericano la mayor probabilidad de expansión y cuentan con pruebas de bioequivalencia porque, entre otras cosas, la FDA las exige como requisito de registro.
Es interesante el acercamiento en función del riesgo que realiza Argentina en su regulación sobre bioequivalencia. En ella se establecieron tres niveles de riesgo sanitario, considerando que existen drogas que deben monitorearse en sangre debido a su estrecha ventana terapéutica, tomando como referencia otras Agencias Sanitarias de países con alta vigilancia sanitaria (Alemania, EE.UU. y Canadá) y teniendo en cuenta la capacidad técnica instalada [16].
La aplicación de una escala de puntuación dio origen a una lista de 17 principios activos, clasificados de alta riesgo sanitario y el grupo farmacológico antirretrovirales (Tabla 1).
Tabla 1. Lista de principios activos que requieren estudios de bioequivalencia en Argentina
|
Fenitoína |
Etosuximida |
|
Digoxina |
Verapamilo |
|
Carbamazepina |
Insulinas |
|
Teofilina |
Levodopa+IDDC |
|
Oxcarbazepina |
Antirretrovirales [17] |
|
Ciclosporina |
Piridostigmina |
|
Carbonato de Litio |
Quinidina |
|
Tolbutamida |
Valproato |
|
Isotertinoína |
Warfarina |
La Red PARF, usando una metodología similar a la Argentina estableció una lista de 15 productos, que no incuye antirretrovirales. La red recomienda una exigencia gradual de las pruebas considerando el riesgo sanitario y las capacidades nacionales [18] (Tabla 2).
Aunque los antirretrovirales no cumplen los criterios de selección de productos para la realización de las pruebas de bioequivalencia y varias regulaciones de países de América Latina hayan sido posteriores a la comercialización de los mismos, con lo cual muchos estarían exentos de cumplirla (por ejemplo Brasil), existen opiniones diversas al respecto. La motivación sobre su exigencia puede obedecer más a la necesidad de resolver problemas de calidad percibida, que de importancia clínica.
Tabla 2. Lista de principios activos para adopción gradual de estudios
de bioequivalencia. Red PARF

Otros países definen a qué productos se exige bioequivalencia, según consenso de expertos o guías nacionales. Por ejemplo, Colombia ha establecido 5 productos [19], considerando criterios como toxicidad, margen terapéutico, comportamiento farmacocinético y/o problemas documentados de eficacia terapéutica. Este mecanismo ha sido adoptado por resolución ministerial [20].
La trampa de la intercambiabilidad
La intercambiabilidad, representa la práctica de la prescripción o de la dispensación en la cual un medicamento competidor adquiere el derecho de ser usado en lugar de un medicamento innovador. ¿Cómo y cuándo se adquiere este derecho? En algunos casos con la presentación de las pruebas de bioequivalencia, en otros con la presentación de otras pruebas, en la vida real, ni lo uno ni lo otro.
El asunto es tan complejo que se convierte en una trampa que tiene dos caras: por un lado la segmentación natural del mercado, en la que los usuarios de medicamentos asumen una diferenciación categórica que se traduce en diferenciaciones de precio.
La industria de genéricos ha entendido muy bien los beneficios y riesgos de dicha segmentación y ha establecido las estrategias necesarias para cumplir la regulación de la exigencia de pruebas de bioequivalencia, atendiendo a sus intereses comerciales.
Por ejemplo, el primer estudio de bioequivalencia presentado en Colombia por iniciativa del fabricante, (no por requerimiento sanitario) fue sobre un medicamento que contiene sindenafil. De un total de 12 competidores, éste se convirtió en el único producto bioequivalente competidor del famoso Viagra®, producto líder de la firma Pfizer. La autoridad sanitaria (el INVIMA) declaró que el estudio estaba bien diseñado y realizado, pero no certificó intercambiabilidad, pues esta no esta contemplada en las normas. El productor intentó presentar una demanda judicial sobre la autoridad reguladora por negarse a declarar la intercambibilidad con el innovador, dado que la regulación colombiana “solo” estableció que el estudio de bioequivalencia estaba bien diseñado y realizado [21].
Dado que está por fuera de discusión la poca importancia de sindenafil en el perfil epidemiológico colombiano, se demuestra cómo, el fin de la realización del estudio era usufructuar la inversión realizada y disputar el liderazgo del sindenafil bioequivalente.
La otra cara de la trampa de la intercambiabilidad, es que su generalización se asocia a serias limitaciones técnicas. Veamos:
El diseño metodológico de los estudios de bioequivalencia supone que el intercambio solo debe producirse entre los dos medicamentos evaluados. La bioequivalencia es un ensayo biológico llevado a cabo de acuerdo a un protocolo rígido para excluir tantas variables como sea posible. Sin embargo, en la práctica habitual se presenta intercambio indiscriminado entre los diferentes competidores (Gráfico 1).
Gráfico 1. Intercambiabilidad en la práctica habitual
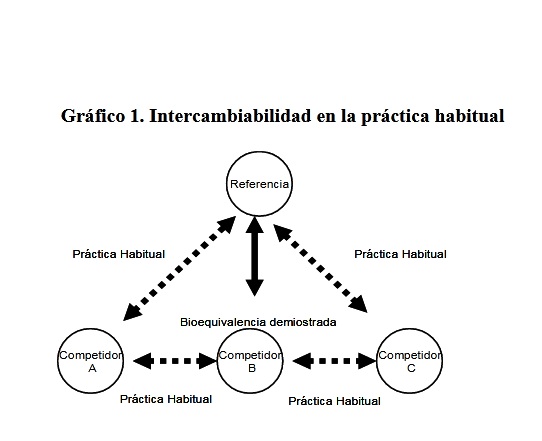
Pero existen demostradas restricciones al intercambio de medicamentos cuando existe alta variabilidad entre sujetos y/o las características de los principios activos. En estos casos se sugiere que no se realice sustitución sin la autorización y monitorización médica [22]. El asunto es que muchos de los productos que requieren bioequivalencia son aquellos que presentan alta variabilidad. Con lo cual, en muchos casos, aún demostrando bioequivalencia, la intercambiabilidad debería restringirse. Adicionalmente, la alta variabilidad puede generar resultados de falsa bio-inequivalencia, dada la muestra reducida (normalmente participan entre 12 y 24 individuos) [23].
Una propuesta de regulación que espera no trasladar el contenido técnico de estos aspectos a los usuarios, es establecer una lista “negativa” de productos que no deben ser sustituidos en ningún sentido (genérico-referencia o referencia-genérico) sin la autorización médica, que corresponde a la misma lista de productos que requieren bioequivalencia. Así. todos los demás productos podrían ser sustituidos libremente [24]. Esta propuesta eliminaría la discusión sobre la intercambiabilidad y permitiría reforzar las iniciativas de sustitución de genéricos en los procesos de prescripción y dispensación.
México optó por un esquema opuesto [25]. En este caso, declara intercambiables los genéricos según el cumplimiento de los tres tipos de pruebas que se describen a continuación:
Pruebas “A”: Cumplimiento de BPF.
Pruebas “B”: Cumplimiento de BPF + perfil de disolución.
Prueba “C”: Cumplimiento de BPF + perfil de disolución + pruebas de bioequivalencia [26].
La porción de productos que requiere bioequivalencia corresponde a cerca del 30% del mercado [27].
Cualquier genérico que se declare intercambiable se comercializa con una etiqueta que destaca las iniciales GI (Genérico Intercambiable) para que el prescriptor y/o el paciente distingan el medicamento en el mercado. Los productos que no han presentado estas pruebas siguen denominándose copias. Hay una lista de genéricos intercambiables conocida como el libro azul, que sigue el mismo concepto del naranja de la FDA y le permite al dispensador, médico o usuario conocer cuáles productos son intercambiables.
Eludir las complicaciones de la intercambibilidad no parece fácil, pero sería inteligente discutir el tema de forma detallada. Desenredar la madeja puede ser, en sí misma, una ganancia.
Mitos y realidades asociados a los estudios de bioequivalencia
Alrededor del tema de la bioequivalencia circulan algunos conceptos erróneos que es bueno desmitificar:
– Mito 1: La equivalencia terapéutica, solo se puede demostrar mediante estudios de bioequivalencia.
La equivalencia terapéutica se puede demostrar también por otros métodos: Ensayos clínicos, estudios farmacodinámicos y pruebas in vitro.
Los estudios in vitro son una alternativa ampliamente estudiada para reducir los costos de los estudios de equivalencia terapéutica y evitar la investigación en humanos. Estos consisten en realizar perfiles de disolución comparativos. Según el esquema de clasificación biofarmacéutica, los productos que son altamente solubles y altamente permeables, no tendrían que realizar estudios en humanos y podrían presentar pruebas in vitro, como demostración de equivalencia terapéutica [28]. Un reciente estudio estableció que el 55% de los medicamentos de la lista de medicamentos esenciales de la OMS cumplen con estas características [29].
– Mito 2: Los genéricos no han comprobado su eficacia, mientras que los productos innovadores si, por eso deben realizar estudios de bioequivalencia.
Las pruebas clínicas y preclínicas se deben presentar una sola vez ante la autoridad sanitaria, en cumplimiento de los principios éticos de la investigación clínica [30]. Por lo tanto, todos los competidores que pretendan comercializar el mismo principio activo solo deben presentar la información farmacéutica o galénica y la legal, lo cual constituye el llamado registro abreviado o sumario, en la mayoría de regulaciones sobre registro de medicamentos.
En el caso de Estados Unidos, cuando un laboratorio desea registrar un medicamento genérico, debe hacerlo siguiendo los procedimientos del Abbreviated New Drug Application (ANDA). La FDA establece que el laboratorio productor no debe repetir los estudios clínicos y preclínicos, pero debe probar la equivalencia terapéutica a través un estudio de bioequivalencia y/u otras pruebas. Como se mencionó en apartados anteriores la aparición de genéricos en muchos países no incorpora este requisito de forma generalizada.
– Mito 3: Los estudios de bioequivalencia se realizan solo para productos genéricos.
La utilidad de los estudios de bioequivalencia no se limita a la comparación entre un producto de referencia y un genérico o competidor, también son realizados por los productores innovadores, especialmente cuando el producto no refleja las mismas características del utilizado en los ensayos clínicos, cuando cambian las características de producción por traslado de país, cambios en el proceso de producción, etc. Algunas regulaciones como la de Estados Unidos, deja a libertad del productor innovador la realización de pruebas de bioequivalencia o biodisponibilidad [31].
– Mito 4: En un estudio de bioequivalencia siempre se compara genérico vs innovador.
No siempre el producto de referencia en el estudio de bioequivalencia corresponde al innovador. La aplicación del sistema de propiedad intelectual corresponde al ámbito nacional y su ampliación nivel global es reciente, por lo tanto muchos países contaron en sus mercados con medicamentos genéricos con patentes vigentes e incluso sin el producto innovador comercializado en su territorio. En estos casos el producto de referencia puede ser el producto líder del mercado u otro producto [32].
Estos mitos, promocionados mediante distintos mecanismos de desinformación, son muestra de la falta de transparencia del mercado farmacéutico y se convierten en una importante barrera de aceptación de los medicamentos genéricos.
Capacidades nacionales y barreras técnicas
El problema de la bioequivalencia no es un problema de mercados “inmaduros” que accepten una producción de genéricos que cumpla con los mismos estándares de los mercados “maduros”. Para producir medicamentos genéricos un laboratorio debe tener el tamaño suficiente para una producción controlada. Incluso el problema no lo constituye el costo de los estudios de bioequivalencia [33].
El problema de fondo está en que las pruebas de bioequivalencia se pueden convertir en un mecanismo de restricción de la competencia, por ejemplo en el marco de los procesos de negociación de acuerdos comerciales en marcha.
En este sentido, debe considerarse que el desarrollo de producción de genéricos en países en desarrollo favorece un desarrollo industrial verdadero, no “clandestino” y representa bienestar y credibilidad ciudadaba. Este asunto, transciende el tema sanitario por que respresenta un marcador del desarrollo productivo, el caso del Brasil es un ejemplo contundente.
En otros términos, el tema de la bioequivalencia cobra importancia en las políticas de genéricos, solo si es un aspecto de naturaleza sanitaria, solamente cuando se propone como una solución a una falla no-controlada del sector productivo y de sus responsabilidades.
Toda esta historia y su evolución, hacen evidente la necesidad de elaboración de guías y documentos de fácil interpretación y consulta. Las declaraciones de la OMS, alrededor del retiro de los antirretrovirales de su lista de precalificación, se convierten en la mejor oportunidad para poner el ojo en el centro del huracán e intentar debatir sin temores un tema que ha sido manejado exclusivamente en espacios técnicos, sin considerar sus implicaciones públicas, sanitarias y políticas.
Reconocimientos:
Las ideas fuerza del documento fueron discutidas ampliamente con Joan Rovira y Gianni Tognoni.
Notas:
(a) Este artículo fue escrito con el interés de propiciar una discusión abierta ante un tema y un momento polémico. Representa opiniones individuales y no compromete a las instituciones a las que los autores pertenecen.
Referencias:
1. WHO Expert Committee on Specifications for Pharmaceutical Preparations. Thirty-fourth report, 1996: 114-154 (WHO Technical Report Series, No.863).
2. www.paho.org/spanish/ad/ths/ev/besp-index.htm ácido valpróico, carbamazepina, ciclosporina, fenitoína.
3. www.paho.org/spanish/ad/ths/ev/besp-index.htm
4. Mensaje lista electrónica e-farmacos, Gerardo E. Valladares A., septiembre de 2004.
5. www.who.int/mediacentre/releases/2004/pr53/en/. Laboratorio Ranbaxy Laboratorios Ltda.: Lamivudina + Estavudina + Nevirapina a dosis fijas, en dos concentraciones diferentes; Lamivudina + Zidovudina a dosis fijas. Laboratorio Cipla: Lamivudina 150mg tabletas y Lamivudina 50mg+Zidovudina 300mg tabletas.
6. WHO Prequalification project. mednet3.who.int/prequal/
7. www.who.int/mediacentre/releases/2004/pr53/en/
8. Posición de las Redes Comunitarias de Latinoamérica y el Caribe frente a la adquisición de medicamentos antirretrovirales genéricos certificados, de marca registrada protegida por patente y copias o similares.
9. Lista electrónica e-drug, mensaje Francisco Rossi, agosto 2004.
10. Lista electrónica diálogos-farmacéuticos, mensaje AIS-Colombia agosto de 2004. Reproducida en la lista electrónica e-fármacos por Francisco Rossi, septiembre de 2004.
11. Productos farmacéuticos de fuentes múltiples: Son productos farmacéuticamente equivalentes que pueden ser o no equivalentes desde el punto de vista terapéutico. Los productos farmacéuticos de fuentes múltiples que son terapéuticamente equivalentes se consideran intercambiables. WHO Expert Committee, Expert Committee on Specifications for Pharmaceutical Preparations. Thirty-fourth report, 1996 (WHO Technical Report Series, No.863).
12. WHO Expert Committee, 1996, op. cit.
13. Tognoni Gianni. Comunicación personal electrónica. Julio de 2004.
14. Giarcovich S. Genéricos, similares y el problema de la intercambiabilidad. Revista de la Sociedad Argentina de Farmacia y Bioquímica Industrial 2001:40(101):10-15.
15. Tognoni Gianni. Comunicación personal electrónica. Septiembre 2004.
16. Requerimiento de estudios de bioequivalencia. ANMAT Nº 3185. Formas farmacéuticas que no requieren BE. Disposición (ANMAT) Nº 2814/02.
17. ANMAT disposición 3311 de 2001.
18. www.paho.org/spanish/ad/ths/ev/besp-index.htm
19. Ácido valproico, carbamacepina, ciclofosfamida, fenitoína y oxacarbamacepina.
20. Ministerio de Salud, Colombia. Resoluciones 1400 y 1890 de 2001.
21. INVIMA. Oficina Jurídica. Ver demanda judicial Pfizer sobre Lafrancol, por violación de los derechos de marca por usar en la etiqueta Eroxim ® bioequivalente a Viagra®.
22. La transitividad de los estudios de bioequivalencia no es absoluta (Si A=B y B=C entonces A=C) y la posibilidad de error tendrá diferentes implicaciones clínicas según los distintos tipos de drogas. Giacovich op. cit.
23. Por ejemplo, para demostrar la bioequivalencia entre dos formulaciones de un principio activo que presenta una variación en la Concentración Sanguínea del 30%, se requeriría incorporar 52 sujetos. Una muestra de este tamaño generaría aumento de los costos y fuertes cuestionamientos éticos. Boddy et al. An approach for widening the bioequivalence acceptance limits in the case of highly variable drugs. Pharmaceutical Research 1995;12(12): 1865-1868.
24. En Colombia existe una propuesta de reglamentación que sugiere incluir fenitoina, carbamacepina, y digoxina. Otros agregan a estos ácido valpróico digoxina, warfarina, litio y teofilina.
25. Dirección General de Insumos para la salud. Norma Oficial Mexicana 177-SSA1-1998.
26. Resaltar que, este caso refleja cómo la prueba de bioequivalencia tiene sentido solo si se realiza con productos cuyo fabricante cumpla con las Buenas Prácticas de Manufactura.
27. Meiuxierio F. Subdirector de Evaluación de Medicamentos. Comisión Federal para la Protección contra Riesgos Sanitarios. Estados Unidos de México. Trabajo presentado en la Reunión sobre Bioequivalencia, 15-16 de abril de 2004, Lima, Perú.
28. FDA. Op cit. 2002.
29. Nehal et al. Molecular properties of WHO Essential Drugs and Provisional Biopharmaceutical Classification. Molecular Pharmaceutics. American Chemical Society 2004; 1(1): 85-96.
30. Asociación Médica Mundial. Principios éticos para las investigaciones médicas en seres humanos. Edimburgo. Helsinki, Finlandia; Junio 1964.
31. Food and Drug Administration. Waiver in vivo BA and BE studies for immediate-released solid oral dosage forms based on biopharmaceutics classification system 2002. Guidance for Industry. Rockeville, MD: FDA; 2002.
31. Giarcovich op. cit.
32. WHO. Technical Report Series, No 902, Annex 1 Guidance on the selection of comparator pharmaceutical products for equivalence assessment of interchangeable multisource (generic) products. Geneva; 2002.
33. El costo de un estudio oscila entre los US$15.000 y 40.000 y depende del principio activo, dosis y forma farmacéutica. Dato suministrado por una institucion especializada en ensayos clinicos. Julio de 2004.