Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Investigaciones
El destino de los estudios de postcomercialización de la FDA (The fate of FDA postapproval studies)
Steven Woloshin, Lisa M. Schwartz, Brian White, Thomas J. Moore
N Engl J Med 2017; 377:1114-1117
DOI: 10.1056/NEJMp1705800
Traducido por Salud y Fármacos
Tanto el Congreso como la FDA han intentado acelerar la disponibilidad de los medicamentos nuevos permitiendo que los patrocinadores esperen a resolver muchas preguntas sobre su seguridad y sus beneficios hasta después de haber recibido el permiso de comercialización. Como resultado, la mayoría de las cartas de aprobación requieren estudios de fase 4 para abordar cuestiones tales como dosis óptimas, posibles efectos secundarios a largo plazo y uso en pediatría, o para confirmar el beneficio clínico de los medicamentos que reciben aprobación condicional en base a evidencia preliminar.
En respuesta a las críticas generalizadas sobre la laxa supervisión de los estudios post comercialización y el hecho de que muchos de estos estudios se quedan inconclusos, el Congreso, a través de la Ley de Enmiendas de la FDA de 2007 (FDAAA), otorgó poder adicional a la FDA para exigir que las compañías los completen. Antes de esa ley, la FDA no tenía autoridad estatutaria específica para ordenar estudios post comercialización; en su lugar condicionaba su aprobación al establecimiento de “compromisos” con el patrocinador. Según la FDAAA, la agencia puede establecer requisitos y compromisos cuando aprueba un medicamento (o más tarde, si surge nueva información de seguridad). Además, la FDAAA autorizó a la FDA a especificar cuándo se deben alcanzar ciertos hitos del estudio, y a emitir multas o cancelar el permiso de comercialización por incumplimiento. La FDA también debe emitir informes anuales sobre el estado de los estudios postcomercialización.
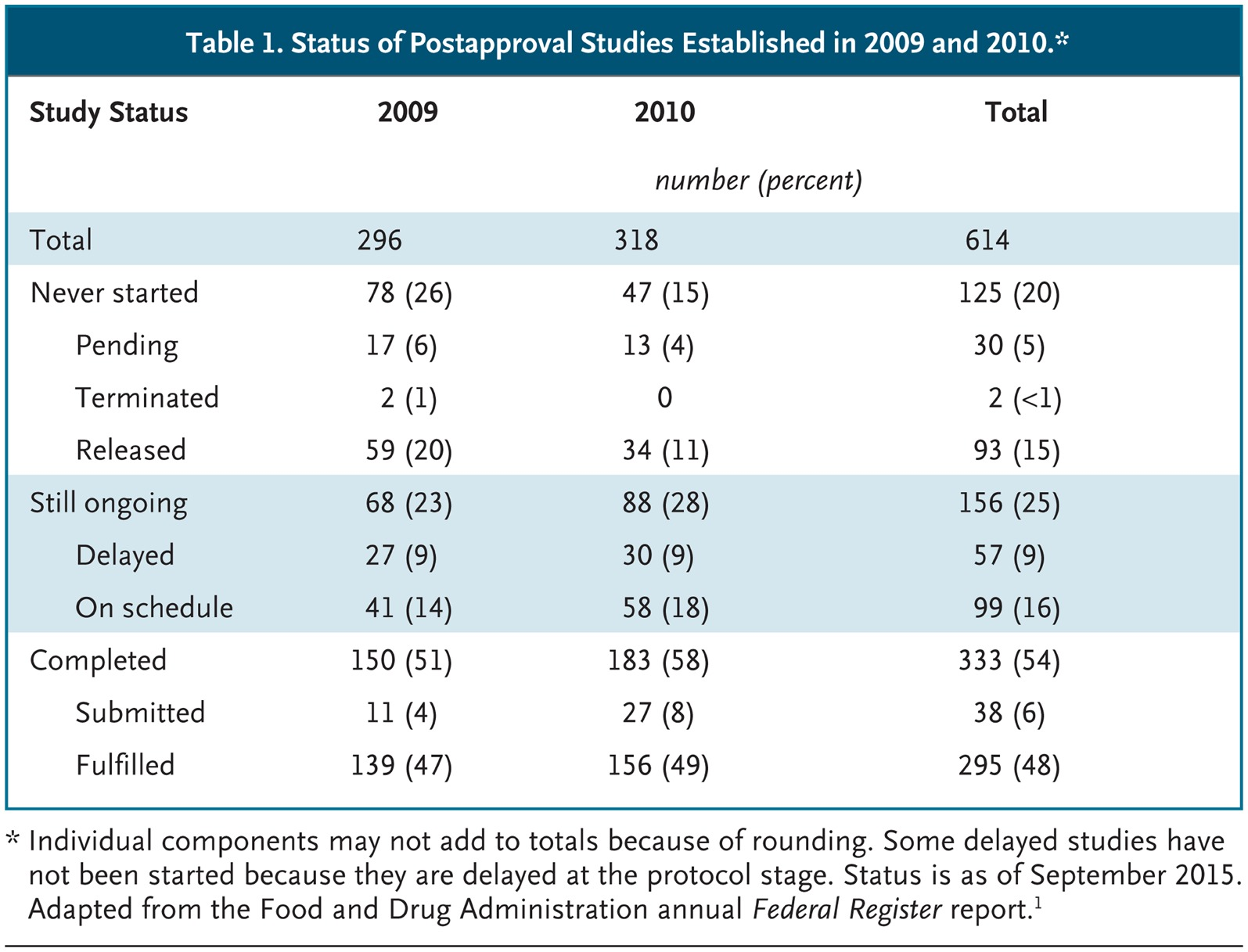
Para evaluar los efectos de la legislación de 2007, utilizamos el último informe de situación del Registro Federal para evaluar 614 requisitos y compromisos postcomercialización que se impusieron en 2009 y 2010, los primeros años cubiertos por el informe (ver Cuadro 1) [1]. Después de entre 5 y 6 años, el 20% de los estudios postcomercialización no habían comenzado, el 25% estaban retrasados o en curso, y el 54% se habían terminado. Analizamos la base de datos de la FDA relacionada con los requisitos y compromisos postcomercialización para obtener ejemplos de los estudios inconclusos (ver Cuadro 2) [2].
Los estudios inconclusos se clasificaron en varias categorías. El 16% estaban en curso y avanzaban según lo programado. Otro 5% también se encontraban dentro del cronograma, pero se clasificaron como pendientes porque no habían inscrito a ningún paciente. Aunque cumplir el calendario es tranquilizador, desde nuestra perspectiva, algunos cronogramas que ha especificado la FDA son demasiado generosos. Por ejemplo, el cronograma de un estudio de eficacia y seguridad de un año para evaluar los efectos de Welchol (colesevelam) en la diabetes tipo 2 en niños de 10 a 17 años, tenía un plazo de 6 años; y se otorgó una extensión adicional de 4 años.
Sin embargo, algunos estudios post comercialización estaban atrasados, lo que significa que no habían cumplido con la fecha límite especificada por la FDA, ya fuera que no habían presentado el protocolo o finalizado el ensayo. En general, el 9% de los estudios postcomercialización se atrasaron. Por ejemplo, la FDA exigió un ensayo clínico para evaluar el riesgo de prolongación del intervalo QT en los pacientes tratados con Suboxone (buprenorfina y naloxona). Aunque el diseño de dichos estudios está altamente estandarizado y se monitorea a los participantes durante un período limitado (por lo general de unos días a un mes), el patrocinador tuvo más de 1 año para enviar el protocolo del estudio y 5 años para completarlo. Sin embargo, en julio de 2017, aparentemente, todavía no había presentado el protocolo final. Según la web de la FDA, el patrocinador “actualmente está en proceso de finalizar la revisión del protocolo para adecuarlo a las recomendaciones de la FDA, y entregará la nueva versión para su revisión antes de implementarlo”.
La FDA exigió un registro para investigar una señal de riesgo de cáncer de tiroides con el uso de Victoza (liraglutida), un medicamento ampliamente recetado para la diabetes, y también se retrasó. El protocolo vencía en julio de 2010, pero al parecer en julio de 2017 todavía no se había recibido. Siete años después de que la FDA estableciera el requisito, aparentemente el registro no se había iniciado.
El caso de Folotyn (pralatrexate) es un ejemplo de atraso en cumplir con los requisitos de postcomercialización cuando se trata de estudios esenciales para evaluar beneficios. El medicamento fue aprobado condicionalmente para el linfoma recurrente o resistente de células T periféricas por la vía de aprobación acelerada, que permite a la FDA aprobar medicamentos para afecciones graves para las cuales hay pocas opciones de tratamiento en base a evidencia preliminar limitada, pero se requieren ensayos posteriores para confirmar el beneficio [3]. El pralatrexato se aprobó en base a los resultados de un ensayo en un solo grupo de 111 pacientes en el que el 26% tuvo una respuesta completa o parcial, y el 44% experimentó un evento adverso grave. Cuando la FDA aprobó el medicamento, se requirieron dos ensayos aleatorizados para confirmar el beneficio clínico, definido como una mayor supervivencia libre de progresión (aunque esa medida no se relaciona de manera consistente con la supervivencia general). Siete años más tarde, un ensayo había incumplido los plazos para dos hitos y había inscrito a 30 pacientes; el otro todavía estaba en curso. La FDA también requirió dos estudios adicionales postcomercialización para cumplir con los requisitos de aprobación acelerada; ambos estaban atrasados. En consecuencia, el etiquetado de pralatrexate aún advierte a los prescriptores que el beneficio clínico no ha sido demostrado.
La FDA clasificó el 15% de los estudios postcomercialización inconclusos como “Liberado”, lo que significa que la FDA liberó al patrocinador de la obligación de realizar el estudio porque “o ya no es viable o ya no brindará información útil”. Algunos de estos estudios podrían haberse completado y podrían haber proporcionado información útil. Por ejemplo, al aprobar Stelara (ustekinumab), la FDA exigió un estudio de lactancia para determinar si, al utilizarlo para tratar la psoriasis en placas de moderada a grave o la artritis psoriásica, pasaba a la leche materna, y para evaluar posibles efectos adversos en lactantes. La FDA posteriormente eximió al patrocinador de hacer el estudio, pero no explicó por qué. Cuando se estableció el estudio, la FDA señaló que se podría hacer en un subgrupo de mujeres utilizando un registro de mujeres embarazadas que se crearía como otro requisito separado de postcomercialización. El estudio podría proporcionar información importante, ya que una buena proporción de mujeres con psoriasis están en edad fértil.
El ritmo lento e irregular de los estudios postcomercialización contrasta marcadamente con los plazos cortos y rígidos, y otros métodos abreviados que se utilizan para acelerar el registro [4]. El acercamiento de la FDA a los estudios postcomercialización ha mejorado en la última década: ha eliminado gran parte del retraso acumulado antes de que se aprobara FDAAA, ha establecido cronogramas y creado una base de datos pública para el seguimiento de los estudios. Sin embargo, creemos que hay que hacer algo más.
En primer lugar, la FDA podría hacer más para garantizar que los patrocinadores cumplen los cronogramas de los estudios postcomercialización. A fines de 2015, solo se habían completado la mitad de los estudios solicitados en 2009 y 2010, y algunas empresas incluso omitieron entregar los informes de actualización anual que se requieren. Según el último informe del Registro Federal, que incluye datos de 2015, el 13% de los informes anuales de actualización se presentaron con al menos dos meses de retraso y 19% no fueron recibidos [1]. La FDA podría usar el poder que le otorga la FDAAA para imponer multas u otras sanciones a los patrocinadores que no cumplan los plazos. Hasta donde sabemos, nunca ha impuesto tales multas [5].
En segundo lugar, aunque el Congreso presiona a la FDA para que reduzca el número de requisitos postcomercialización que no se cumplen, debe tener mucho cuidado en liberar a los patrocinadores de la obligación de realizar estudios previamente requeridos. Tales decisiones son apropiadas cuando ya no se necesita el estudio, por ejemplo, cuando ya se ha respondido la pregunta central, o cuando surgen problemas insuperables de viabilidad. Desafortunadamente, las razones de la FDA para eliminar los requisitos no se hacen públicas. Agregar esta información a su base pública de datos postcomercialización mejoraría la transparencia.
Finalmente, creemos que la FDA debería considerar cronogramas más cortos. Una estrategia sería reducir el tiempo entre la aprobación del medicamento y el inicio de los estudios postcomercialización, identificando los requisitos postcomercialización tan pronto como sea posible en el proceso de revisión. Los plazos razonables para realizar el estudio clínico se pueden estimar a partir de la base de datos ClinicalTrials.gov, que enumera las fechas de inicio y finalización de la mayoría de los ensayos clínicos con medicamentos. Muchos estudios farmacocinéticos y farmacodinámicos, por ejemplo, pueden realizarse en meses en lugar de años.
Con el aumento del uso de los procesos acelerados de revisión de la FDA y el compromiso expreso del presidente Donald Trump de acelerar el proceso de aprobación “lento y oneroso” de la FDA, es probable que la aprobación de medicamentos sea cada vez más rápida y dependa de estándares de evidencia más flexibles. Será crucial garantizar que las preguntas importantes que no se responden en el momento de la aprobación también se resuelvan lo más rápidamente posible.
Referencias