Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Ensayos Clínicos
Gestión de los Ensayos Clínicos, Metodología y Conflictos de Intereses
Observatorio de ensayos: Tendencias en la complejidad del diseño de ensayos clínicos (Trial watch: Trends in clinical trial design complexity)
Getz KA, Campo RA, Tufts Center for the Study of Drug Development
Nature Reviews Drug Discovery 2017; 16:307 doi:10.1038/nrd.2017.65
Traducido por Salud y Fármacos
Durante los últimos 15 años, los retos de medir la seguridad y la eficacia de los fármacos que se están investigando para combatir enfermedades crónicas, difíciles de tratar o enfermedades raras en subpoblaciones de pacientes más estrictamente definidas, han ampliado el alcance de los ensayos clínicos y la carga de ejecutarlos. Otros factores que afectan al diseño del protocolo incluyen la captura de más medidas de resultado auto reportadas por el paciente y la recopilación de datos comparativos de eficacia y biomarcadores. Aquí ofrecemos nuevos datos de referencia sobre la complejidad de los ensayos, con el objetivo de que los patrocinadores del desarrollo de fármacos puedan comparar sus propias prácticas organizativas e informen a los profesionales de la investigación clínica sobre cómo ha ido evolucionando su diseño de protocolos.
El análisis se basa en 9.737 protocolos de ensayos clínicos que recibieron la aprobación del comité de ética entre 2001 y 2015, y han sido extraídos de la base de datos PICAS de Medidata, que contiene protocolos e información detallada sobre los contratos con los centros de investigación de más de 170 compañías farmacéuticas y biotecnológicas globales (76% fueron proporcionados por las grandes empresas y 24% por empresas medianas y pequeñas (ver información adicional en S1 [cuadro] del documento original para más detalles). Se evaluaron y compararon elementos del diseño de los ensayos en dos períodos de tiempo – 2001-2005 y 2011-2015, con 10 años de separación para caracterizar tendencias. Este diseño permitió ampliar el periodo del análisis para obtener las ideas más significativas, y reducir el efecto de los ensayos que se salen de lo normal (outliers) en un año determinado.
Los elementos de diseño que se incluyeron estaban relacionados con la factibilidad de ejecución – incluyendo el número de procedimientos realizados, el número planificado de visitas de los voluntarios, el esfuerzo requerido para llevar a cabo los procedimientos y el costo por visita del voluntario del estudio.
Los resultados de este análisis muestran que los elementos de diseño de protocolo que se asocian a su ejecución han aumentado rápidamente. El número medio de procedimientos distintos por protocolo aumentó significativamente para las fases I, II y III, especialmente entre los protocolos de fase II y III (Figura 1a). La frecuencia con la que se realizó cada procedimiento creció a un ritmo aún más rápido, lo que condujo a un mayor crecimiento en el número medio de procedimientos totales. Durante este período, sin embargo, el número medio de visitas planificadas por voluntario creció a un ritmo mucho más modesto, lo que resultó en más procedimientos realizados por visita y una mayor carga sobre el voluntario.
Figura 1: Tendencias en la complejidad y los costos de los ensayos clínicosa | Tasas de crecimiento de elementos en el diseño de protocolos entre 2001-2005 y 2011-2015. b | Costo por visita del participante para los mismos dos periodos. El aumento de la complejidad del protocolo ha compensado el ahorro por la eficiencia en los procedimientos y los avances tecnológicos. Vea información adicional S1 (cuadro) en el documento original para más detalles.
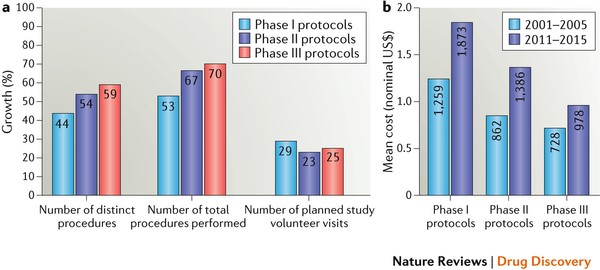
Los protocolos de Fase I realizados en el período 2011-2015fueron los más complejos, tuvieron la media más alta en número de procedimientos distintos (36) y de procedimientos totales (253). Los protocolos de Fase III registraron el mayor crecimiento relativo en el total de procedimientos llevados a cabo, aumentando en un 70%, de una media de 110 procedimientos en 2001-2005 a 187 en 2011-2015 (Figura 1a). En 2001-2005 se realizaron un promedio de 22 procedimientos distintos por protocolo de fase III, en comparación con 35 en 2011-2015 – un aumento del 59%. El número medio de visitas planificadas de los voluntarios aumentó un 25%, de 12 visitas por protocolo en 2001-2005 a 15 visitas por protocolo en 2011-2015.
El trabajo necesario para administrar los protocolos de fases I, II y III en los centros de investigación también aumentó sustancialmente en el período de 10 años (información complementaria S1 [cuadro] en el artículo original), al igual que el costo medio por voluntario por visita (figura 1b). Aunque los costos de muchos procedimientos, como las pruebas de sangre, han disminuido durante la última década, el costo total por visita del voluntario ha aumentado considerablemente debido al aumento del número total de procedimientos realizados. Los estudios de Fase II registraron el mayor incremento en el costo nominal medio por visita (61%), seguido de cerca por los estudios de fase I (49%). Los estudios de Fase III mostraron un crecimiento más modesto en el costo promedio por visita, con un incremento del 34% en el período de 10 años.
Estos hallazgos son sorprendentes dada la existencia de investigación vinculando la complejidad del protocolo con ciclos más largos, un mayor número de enmiendas de protocolo y menores tasas de reclutamiento y retención de pacientes (por ejemplo, Contemp. Clin. Trials 28, 583-592, 2007). La recopilación de datos clínicos excesivos e innecesarios también puede comprometer la integridad y el análisis de los datos, resultar en tasas de error más altas, prolongar la duración de los estudios y retrasar las presentaciones a las agencias reguladoras.
Un número creciente de empresas farmacéuticas y de biotecnología y organizaciones de investigación por contrato (CROs) han adoptado medidas para optimizar sus diseños de protocolos a fin de mejorar la viabilidad, aliviar la carga para el voluntario y el centro de investigación, reducir el número de enmiendas no planificadas y no presupuestadas y reunir datos clínicos más significativos. Estas iniciativas incluyen comités de revisión de protocolos, prácticas de autentificación de protocolos que conecten los procedimientos con las medidas de impacto primarias y secundarias clave, plantillas comunes de redacción de protocolos y solicitudes de retroalimentación a los pacientes y al personal de investigación sobre los proyectos de diseño de protocolos antes de su aprobación y ejecución. Los estudios realizados en el Centro Tufts para el Estudio del Desarrollo de Medicamentos aún no han detectado un impacto medible de estas iniciativas en toda la industria, aunque los primeros informes anecdóticos indican que estas iniciativas están comenzando a producir reducciones en el número de enmiendas al protocolo y en la carga investigativa (Ther. Innov. Regul. Sci. 47, 651 – 655, 2013).