Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Introducción
El objetivo de esta guía es aclarar a los patrocinadores de medicamentos y a otras partes interesadas cómo la FDA incorpora las consideraciones sobre los beneficios, los riesgos y las opciones de gestión de riesgos de un medicamento en ciertas decisiones regulatorias, previas y posteriores a la comercialización.
Esto afecta a las solicitudes de comercialización de nuevos medicamentos (NDA) presentadas en virtud de la sección 505(c) de la Ley Federal de Alimentos, Medicamentos y Cosméticos (Federal Food, Drug, and Cosmetic Act o FD&C Act), así como a las solicitudes de licencia de biológicos (BLA Biological Licence Application) presentadas en virtud de la sección 351(a) de la Ley de Servicios de Salud Pública (Public Health Service Act o PHS Act).
Esta guía empieza discutiendo los aspectos más importantes que el Centro de Evaluación e Investigación de Medicamentos (CDER) y el Centro de Evaluación e Investigación de Productos Biológicos (CBER) tienen en cuenta en las evaluaciones beneficio-riesgo, incluyendo cómo se pueden utilizar los datos de la experiencia de los pacientes en la evaluación beneficio-riesgo.
A continuación, se describe cómo los patrocinadores pueden influir en la evaluación beneficio-riesgo de la FDA a través del diseño y la realización de un programa de desarrollo, así como en la forma de presentar la información sobre el beneficio y el riesgo en la solicitud de comercialización. También discute las oportunidades de interacción entre la FDA y los patrocinadores para discutir las consideraciones beneficio-riesgo en relación con el desarrollo de una NDA o BLA.
Esta guía concluye con consideraciones adicionales sobre las evaluaciones beneficio-riesgo que informan la toma de decisiones regulatorias en el entorno post-comercialización. (Nota: estas interacciones entre la industria y la FDA suelen darse al final de los ensayos clínicos de fase 2).
Esta guía se refiere a las evaluaciones beneficio-riesgo que se realizan para apoyar ciertas decisiones regulatorias sobre los NDAs o BLAs, desde la aprobación previa a la comercialización hasta el entorno post-comercialización. Esto incluye las decisiones relativas a cualquier requisito reglamentario para la aprobación, como la inclusión de una advertencia de caja en el etiquetado /ficha técnica cuando se apruebe el producto, los requisitos y compromisos de completar estudios posteriores a la comercialización y las estrategias de evaluación y mitigación de riesgos (REMS). Estas decisiones regulatorias se establecen de acuerdo con las autoridades y los criterios legales y regulatorios específicos que sean aplicables. Esta guía toca algunos de estos principios, pero no intenta enumerarlos o abordarlos todos.
Esta guía no aborda directamente otras decisiones regulatorias que se pueden dar a lo largo del ciclo del desarrollo de un fármaco, como las decisiones relativas a los primeros ensayos en humanos de un nuevo fármaco en investigación (IND) o las solicitudes de acceso ampliado, que también pueden requerir que la FDA considere la información sobre los beneficios y riesgos de un fármaco en investigación o comercializado para el uso propuesto. Sin embargo, los conceptos discutidos en esta guía pueden ser relevantes para otros tipos de decisiones.
La Agencia desarrolló esta guía adhiriéndose a los objetivos de la sexta autorización de la Ley de Tarifas para Usuarios de Medicamentos de Venta con Receta (PDUFA VI), Título I de la Ley de Reautorización de la FDA de 2017, y los requisitos de la sección 3002(c)(8) de la Ley de Curas del Siglo XXI para emitir guías relacionadas con el uso de datos relevantes de la experiencia de los pacientes e información relacionada para informar la toma de decisiones regulatorias. Esta guía se basa en, y es consistente con, la orientación del Consejo Internacional de Armonización (ICH) para la industria M4E(R2): El documento técnico común (CTD)-Eficacia (ICH M4E(R2)) (julio de 2017).
En general, las guías de la FDA no son obligatorias desde el punto de vista legal. Las guías describen el pensamiento actual de la Agencia sobre un tema y se deben considerar como recomendaciones, a menos que se citen requisitos reglamentarios o legales específicos. El uso de la palabra “debería” en las guías de la Agencia significa que se sugiere o recomienda algo, pero no se exige.
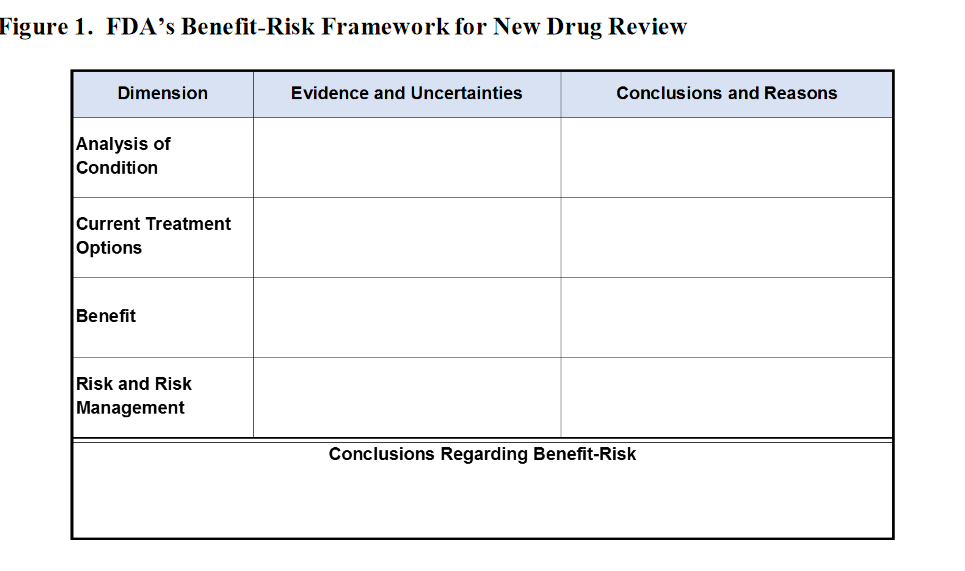
Nota de Salud y Fármacos. La Guía sugiere presentar la información riesgo-beneficio en el siguiente cuadro:
Clinical Leader añade la siguiente información [1]. Para determinar el balance beneficio-riesgo se suele hacer una evaluación cualitativa, que sospesa los datos y la información sobre los beneficios y los riesgos del medicamento, y considera las incertidumbres dentro de un contexto terapéutico y normativo específico. Hacer análisis riesgo-beneficio durante las primeras etapas de desarrollo puede aportar beneficios.
La planificación del análisis beneficio-riesgo incluye la identificación de los pacientes, la recogida de datos, la selección de los criterio primarios de valoración de su eficacia, el uso de controles activos para establecer el perfil beneficio-riesgo, la demostración del beneficio en una subpoblación específica, el tamaño de la muestra y la duración requerida del ensayo clínico, la evaluación de un posible acontecimiento o acontecimientos adversos y las actividades de mitigación del riesgo para prevenir o vigilar los acontecimientos adversos graves previstos. Este plan debe abarcar todo el ciclo de desarrollo del medicamento.
La evaluación beneficio-riesgo de la FDA para la aprobación de nuevos medicamentos incorpora amplias consideraciones de salud pública para la población de pacientes a la que se dirige y otras, como los riesgos relacionados con el mal uso, la exposición accidental o la transmisión de enfermedades. Comprende una evaluación multidisciplinar de la ciencia y la medicina, específica para cada caso, que tiene en cuenta el contexto terapéutico en el que se utilizará el fármaco frente a los tratamientos disponibles, las pruebas presentadas en la solicitud previa a la comercialización y/o generadas durante la vigilancia posterior a la comercialización, las incertidumbres sobre los beneficios y los riesgos del fármaco y las opciones normativas de la FDA para gestionar los riesgos y reducir las incertidumbres.
Referencia