Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
AGENCIAS REGULADORAS Y POLÍTICAS
Investigaciones
۞ Razones científicas y administrativas para el retraso o el rechazo de la aprobación por la FDA de solicitudes iniciales de nuevos medicamentos, 2000-2012
(Scientific and regulatory reasons for delay and denial of FDA approval of initial applications for new drugs, 2000-2012)
Sacks LV, Shamsuddin HH, Yasinskaya YI, Bouri K, Lanthier ML, Sheman RE
JAMA 2014; 311(4):378-84
Traducido por Jose Manuel López Navarro y Carlos García-Navas Vidal (estudiantes de 5º curso de farmacia UMH), revisado por Emilio Pol Yanguas (PharmD, PhD)
Importancia: Algunas solicitudes de aprobación de nuevos medicamentos fracasan debido a un inadecuado rendimiento del medicamento y otras no son aprobadas debido a que la información remitida a la FDA es inadecuada para tomar tal decisión. Volver a enviar la solicitud rechazada es costoso, retrasa la puesta en el mercado y la disponibilidad del nuevo medicamento para los pacientes.
Objetivo: Identificar las razones por las que la FDA rechaza o demora la aprobación de un nuevo medicamento.
Diseño, lugar y participantes: Se realizó una revisión y extracción de datos de los documentos de la FDA de forma retrospectiva. Hemos examinado todas las primeras solicitudes de aprobación de medicamentos remitidas a la FDA entre 2000 y 2012 para nuevas entidades moleculares (NMEs), que son ingredientes activos nunca antes comercializados en EE UU en forma alguna. Utilizando la correspondencia y revisiones de la FDA, hemos investigado las razones por las que las NMEs fracasaron en la obtención de la aprobación de la FDA.
Principales variables resultado y medidas: Razones de la FDA para el retraso o el rechazo de la aprobación de comercialización de NMEs.
Resultados: De las 302 solicitudes NMEs identificadas, 151 (50%) fueron aprobadas tras su consideración inicial, y hasta 222 (73,5%) fueron aprobados finalmente. Setenta y una solicitudes requirieron de una o más reconsideraciones antes de ser aprobadas, con una mediana de 435 días de demora tras el fracaso en la consideración inicial. De las solicitudes de aprobación fracasadas inicialmente, 24 (15,9%) incluían incertidumbres respecto a la selección de la dosis, 20 (13,2%) selección de variables a medir en el estudio que no reflejaba adecuadamente un efecto clínicamente significativo, 20 (13,2%) resultados inconsistente cuando se examinaron diferentes variables medidas, 17 (11,3%) resultados inconsistentes cuando se compararon diferentes ensayos o lugares del estudio, y 20 (13,2%) pobre eficacia cuando se compara con un patrón de cuidados. La frecuencia de deficiencias de seguridad fue similar entre aquellos medicamentos que no se aprobaron nunca en comparación con aquellos cuya aprobación se demoró (43 de 80 [53,8%] que nunca se aprobaron vs 37 de 71 [52,1%] eventualmente aprobados, diferencia 1,7% [IC95, -14,86% a 18,05%]; p=0,87). Sin embargo, las deficiencias en eficacia fueron significativamente más frecuentes entre los medicamentos que nunca llegaron a aprobarse que entre aquellos con aprobación demorada (61 de 80 [76,3%] que nunca se aprobaron vs 28 de 71 [39,4%] con aprobación demorada; diferencia 36,9% [IC95%, 20,25 a 50,86], p<0,001).
Conclusión y relevancia: Varias deficiencias potencialmente prevenibles, incluyendo fracaso en seleccionar dosis óptimas del medicamento e inadecuadas variables resultado a medir, son responsables de retrasos significativos en la aprobación de nuevos medicamentos. Comprender las razones fracasos previos ayuda a mejorar la eficiencia del desarrollo clínico de nuevos medicamentos.
Introducción
El recorrido desde el descubrimiento de un producto medicinal hasta el mercado es típicamente largo y costoso. El intervalo entre las pruebas clínicas iniciales y la aprobación del producto se ha estimado en un promedio de 8 años [1] y solo 1 de cada 6 fármacos que inician ensayos clínicos obtienen finalmente la aprobación por la FDA[2]. Para obtener la aprobación para la comercialización de un nuevo medicamento, los promotores deben proporcionar evidencia sustancial de seguridad y eficacia para la indicación propuesta [3]. En un número de solicitudes de comercialización fracasadas las deficiencias son adecuadamente dirigidas en la reformulación de la solicitud dando lugar a un retraso en la aprobación, mientras que en otros casos nunca se llega a aprobar la puesta en el mercado. El rechazo de medicamentos es crítico para prevenir la comercialización de productos inefectivos o dañinos. Sin embargo, muchos medicamentos no son aprobados, no a causa de que sean inefectivos o inseguros, sino debido a que la información es inadecuada para hacer estas afirmaciones. Los fracasos y los retrasos que se producen en las etapas finales del desarrollo de un producto afectan a la disponibilidad de productos innovadores por los pacientes e incrementan los costos del desarrollo de medicamentos [4]. Para evitar deficiencias prevenibles en los estadios tardíos del desarrollo de medicamentos y sus consecuencias negativas, es importante comprender la naturaleza de estas deficiencias.
Hemos revisado las solicitudes de comercialización de nuevas entidades moleculares (NMEs) remitidas a la FDA para caracterizar las razones científicas y administrativas por las que su aprobación fue rechazada o demorada.
Método
Hemos examinado todas las solicitudes de comercialización de NMEs terapéuticas que fueron remitidas al “Centro para la investigación y evaluación de medicamentos” (CDER) por primera vez entre el 1-10-2000 y 30-09-2012. Esto no representa a todas las solicitudes de comercialización de medicamentos a la FDA y no incluye las correspondientes a medicamentos genéricos, las solicitudes suplementarias (nuevas indicaciones), ni solicitudes de productos biológicos.
Las solicitudes múltiples de una misma NME en varias formas de dosificación (por ejemplo, comprimidos suspensión, inyectables) y las solicitudes múltiples para diferentes indicaciones fueron evaluadas solo una vez. Los productos diagnósticos no terapéuticos, tales como agentes radiocontrastes, y las solicitudes de medicamentos retiradas por el patrocinador antes de que la FDA realizara ninguna acción, fueron excluidas.
Todas las nuevas solicitudes de comercialización rechazadas cuando fueron remitidas por primera vez a la FDA fueron consideradas fracasos. Hemos extraído la información procedente de las “cartas de acción” de la FDA (respuestas oficiales de la FDA a las solicitudes fracasadas) y las revisiones tanto internas como las públicamente disponibles, así como de las correspondencias que describen las deficiencias identificadas por la FDA. La información públicamente disponible sobre los medicamentos rechazados y aprobados está colgada en el sitio web de la FDA [5] y en las transcripciones de las reuniones de los comités asesores [6].
Hemos agrupado las deficiencias en 4 categorías primarias: 1) eficacia, 2) seguridad, 3) química, fabricación y control (CMC), y 4) prospecto y etiquetado. Estas categorías son individualmente tratadas en solicitudes renovadas de aprobación, las revisiones de la FDA y las cartas de acciones, proporcionando un marco de trabajo lógico para el análisis de las deficiencias. Deficiencias de eficacia hace referencia a aquellas en las que el efecto pretendido del medicamento no se demuestra de manera satisfactoria, o la naturaleza o el tamaño del efecto pretendido no fueron satisfactorios como para permitir la aprobación del medicamento. Las deficiencias de seguridad hacen referencia a aquellas para las cuales la caracterización del riesgo de sucesos adversos o la naturaleza o gravedad de los sucesos adversos observados fueron insatisfactorias para permitir la aprobación del medicamento. Dentro de cada una de estas categorías hemos documentado más detalladamente las razones por las que la seguridad o la eficacia no estuvieron satisfactoriamente demostradas. Las deficiencias químicas, de fabricación y control, fueron aquellas en las cuales no pudo asegurarse la adecuada calidad física del producto farmacéutico. Las deficiencias de prospecto y etiquetado fueron aquellas en las que el prospecto y la etiqueta fracasan en representar adecuadamente la información necesaria para los consumidores en el mercado.
Hemos definido incertidumbre o desacuerdos en la dosis como la incapacidad de determinar una dosificación adecuada para indicar en el prospecto. Esto es generalmente el resultado de una inadecuada exploración de la dosis e incluye solicitudes de comercialización con datos inadecuados de eficacia y seguridad para la dosis propuesta, datos conflictivos de eficacia y seguridad cuando la misma dosis se ha empleado en diferentes estudios, y efectos tóxicos relacionados con la dosis cuando una dosis menor parece potencialmente efectiva. A diferencia de deficiencias tales como inadecuado objetivo final, que categorizamos primariamente como deficiencias en la demostración de la eficacia más que de la seguridad, la imprecisión de dosis inevitablemente afecta a la evaluación tanto de la eficacia como de la seguridad del medicamento. Por tanto la duda o desacuerdo en la dosis se trató como deficiencia en ambos, seguridad y eficacia.
Hemos recogido todas las deficiencias citadas en la “carta de acción” para todas las formas de dosificación del fármaco que hayan tenido algún potencial papel en la decisión reguladora, omitiendo deficiencias menores (tales como problemas de envasado, especificaciones de fabricación subsanables, y preocupaciones clínicas carentes de gravedad) frecuentemente abordadas después de la aprobación. La decisión de incluir una deficiencia en el análisis se hizo por consenso del equipo revisor basándose en lo declarado en la “carta de acción”.
Muchos medicamentos no aprobados tras la consideración inicial (fracasos en el primer ciclo) fueron vueltos a remitir para su consideración por los patrocinadores después de reconducir las deficiencias. Comparamos las deficiencias en solicitudes de comercialización que eventualmente fueron aprobadas hasta el 30 de junio del 2013 (aprobación demorada), con aquellas en las solicitudes que no fueron aprobadas durante el periodo de nuestro estudio (nunca aprobadas) para identificar aquellas que fueron corregidas después de una solicitud fracasada y que podrían haberse prevenido si hubieran sido detectadas tempranamente.
Comparamos las tasas de aprobación para medicamentos a los que se les otorga prioridad de revisión con aquellos a los que se les somete a la revisión usual. Las solicitudes de comercialización cualifican para revisión prioritaria si el nuevo medicamento trata una condición grave y parece aportar una mejoría significativa en seguridad o eficacia en relación con las terapias disponibles [7].
Utilizamos estadística descriptiva para representar las tasas de aprobación y frecuencias de las deficiencias específicas. La prueba exacta de Fischer (SAS Institute Inc., versión 9.3) se utilizó para comparar las diferencias en la frecuencia de deficiencias. Consideramos estadísticamente significativos los valores p<0,05 a dos colas.
Resultados
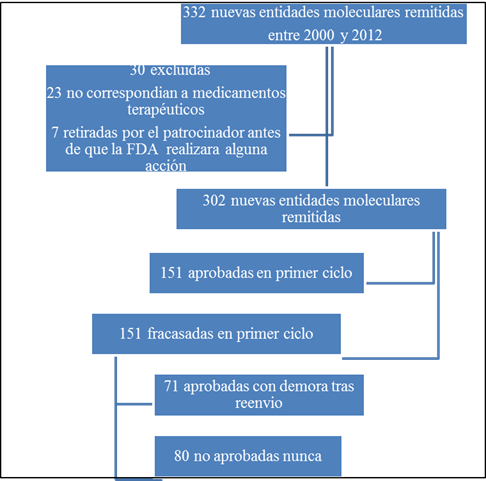
En los 12 años entre el 1 de octubre del 2000 y el 30 de septiembre del 2013 se archivaron en el CDER solicitudes de comercialización de medicamentos de 332 NMEs. Se excluyeron treinta y un medicamentos no terapéuticos (por ejemplo agentes de contraste radiológico) y 7 solicitudes retiradas por el patrocinador antes de que se produjera una acción de la FDA. De las restantes 302 NMEs, 151 (50%) fueron aprobadas tras el primer ciclo de revisión (ver Figura 1).
 |
La tasa de aprobación en primer ciclo fue 72 de 106 (67,9%) para solicitudes a las que se le otorgó revisión prioritaria y 79 de 196 (40,3%) para medicamentos con revisión usual.
La tasa de aprobación varió para las diferentes especialidades médicas (Tabla 1) variando de 72% para medicamentos oncológicos a 31% para medicamentos de neumología y alergología.
Ochenta y siete de los 151 fracasos en primer ciclo (57,6%) volvieron a ser sometidos a revisión para la misma indicación antes del 30 de junio del 2013. De estos, 55 solicitudes (63,2%) fueron aprobadas durante un segundo ciclo de revisiones, 13 (14,9%) durante un tercer ciclo de revisiones, y 3 (3,4%) durante posteriores ciclos de revisión. De los 151 fracasos de primer ciclo, 71 (47,0%) obtuvieron eventualmente aprobación en una mediana de 435 días tras la primera “carta de acción” (rango 47-2374 días). En conjunto, de los 302 medicamentos evaluados, 222 (73,5%) eventualmente lograron la aprobación de comercialización durante el estudio.
Motivos de fracaso de las solicitudes de comercialización de medicamentos
Fracaso en la selección de la dosis. En 24 fracasos de primer ciclo (15,9%) hubo incertidumbre sobre la dosis óptima para maximizar la eficacia y minimizar los riesgos de seguridad. La mayoría de los fármacos para los cuales la FDA recomendó explorar otras dosis, pretendían tratar enfermedades crónicas, tales como desórdenes convulsivos (3 fármacos), dolor e inflamación (2 fármacos), asma (2 fármacos), hipertensión (1 fármaco). Y angina (1 fármaco). Un medicamento antimicrobiano y otro para tratar hemorragias agudas fueron las dos únicas excepciones.
Eficacia. Las razones por las que los productos fracasaron en demostrar su eficacia se recogen en la Tabla 2. Siete medicamentos (7,3%) fracasaron debido a que la población que estudiaron no reflejaba la que probablemente utilizaría el fármaco. Veinte medicamentos (13,2%) fracasaron debido a que las variables resultado medidas en los ensayos clínicos fueron inadecuadas para la aprobación. Las variables resultado inadecuadas incluyeron aquellas de debido a su naturaleza, o el momento en que fueron medidas, fracasaron en capturar un beneficio clínicamente significativo. Ejemplos incluyen medidas de resultados en un momento del tiempo demasiado temprano para mostrar el efecto completo del tratamiento, desacuerdos entre los revisores de la FDA y los investigadores principales en relación con la interpretación de resultado de tratamiento exitoso, ensayos en cáncer que muestran eficacia en la variable resultado supervivencia libre de enfermedad pero no en la supervivencia global, y ensayos con variable resultado de cambio en una medida patológica (por ejemplo, capacidad vital forzada, ácido úrico) para la cual el tamaño del cambio no tiene relación conocida con un beneficio clínico.
| Tabla 1. Frecuencia de aprobación en primer ciclo, según especialidades médicas. | ||
| Especialidad médica | Total de NMEs remitidas | Aprobadas durante el primer ciclo de revisión, Nº (%) |
| Oncología | 61 | 44 (72) |
| Enfermedades metabólicas a | 45 | 21 (47) |
| Neurología / psiquiatría | 42 | 14 (33) |
| Enfermedades infecciosas | 39 | 23 (59) |
| Cardiología | 22 | 7 (32) |
| Oftalmología | 15 | 7 (47) |
| Neumología / alergología | 13 | 4 (31) |
| Gastroenterología | 13 | 9 (69) |
| Urología | 11 | 4 (36) |
| Medicina reproductiva | 10 | 4 (40) |
| Dermatología | 9 | 3 (33) |
| Reumatología / analgesia | 7 | 3 (43) |
| Hematología / hemostasia | 7 | 4 (57) |
| Otros | 8 | 4 (50) |
|
Abreviaturas: NME:: nuevas entidades moleculares a Incluye diabetes mellitus, hiperlipidemia, sobrecarga de hierro, hiponatremia, osteoporosis, síntomas de la menopausia, hiperuricemia, obesidad y enfermedad de Gaucher. |
||
| Tabla 2. Deficiencias en la demostración de eficacia durante el primer ciclo de revisión a | ||||
| Deficiencias de eficacia, Nº (%) | ||||
| Fracasos en el primer ciclo de revisión (n=151) | Aprobación demorada, tras reenvío (n=71) | Medicamentos nunca aprobados durante el estudio (n=80) | ||
| Población | ||||
| Población inadecuada para probar el uso pretendido | 11 (7,3) | 3 (4,2) | 8 (10,0) | |
| Tamaño de la población demasiado pequeño para demostrar eficacia | 4 (2,6) | 0 | 4 (5,0) | |
| Intervención | ||||
| Incertidumbre o desacuerdo sobre la dosis apropiada | 24 (15,9) | 9 (12,7) | 15 (18,8) | |
| Incapacidad para definir el margen de no inferioridad b | 9 (6,0) | 3 (4,2) | 6 (7,5) | |
| Medicación concomitante como factor de confusión | 8 (5,3) | 2 (2,8) | 6 (7,5) | |
| Variable resultado | ||||
| insatisfactoria | 20 (13,2) | 5 (7,0) | 15 (18,8) | |
| Ejecución del estudio | ||||
| Datos perdidos | 3 (2,0) | 0 | 3 (3,8) | |
| Integridad de los datos | 8 (5,3) | 4 (5,6) | 4 (5,2) | |
| Resultado del estudio | ||||
| Resultados inconsistentes en las distintas variables medidas | 20 (13,2) | 6 (8,5) | 14 (17,5) | |
| Resultados inconsistentes en diferentes ensayos o lugares | 17 (11,3) | 3 (4,2) | 14 (17,5) | |
| Eficacia inadecuada en comparación con el cuidado usual | 20 (13,2) | 7 (9,9) | 13 (16,3) | |
|
a En cada solicitud podía haber múltiples tipos de deficiencias b El margen de no inferioridad se determina en función de tamaño del efecto de fármaco control aprobado y la disminución máxima de eficacia en relación con el control que se juzga médicamente aceptable |
||||
| Tabla 3. Número de fármacos con eventos adversos significativos sucedidos durante los ensayos clínicos | |||
| Tipo de evento adversos | Nº a | ||
| Fracasados en el primer ciclo de revisión. (n=151) | Aprobación demorara tras el reenvío (n=71) | Medicamentos nunca aprobados durante el estudio (n=80) | |
| Cardiovascular b | 14 (9,3) | 3 (4,2) | 11 (13,8) |
| Mortalidad global c | 11 (7,3) | 0 | 11 (13,8) |
| Hepático | 9 (5,9) | 2 (2,8) | 7 (8,8) |
| Neuropsiquiátrico | 9 (5,9) | 3 (4,2) | 6 (7,5) |
| Hemostasis | 6 (4,0) | 1 (1,4) | 5 (6,3) |
| Gastrointestinal | 5 (3,3) | 2 (2,8) | 3 (3,8) |
| Interacción medicamentosa | 4 (2,6) | 0 | 4 (5,0) |
| Infección | 4 (2,6) | 3 (4,2) | 1 (1,3) |
| Alergia /inmunología | 4 (2,6) | 1 (1,4) | 3 (3,8) |
| Neoplasia | 4 (2,6) | 2 (2,8) | 2 (2,5) |
| Renal | 3 (2,0) | 3 (4,2) | 0 |
| Musculoesquelética | 2 (1,3) | 2 (2,8) | 0 |
|
a No todas los medicamentos fracasados presentaron efectos adversos significativos. Ver Tablas 2,4 y 5 para otras deficiencias b Incluye acción proarrítmica, efectos sobre la función miocárdica, y sucesos tromboembólicos que afectaron a las coronarias, vasos cerebrales so periféricos. c todas las causas de mortalidad fueron mayores en el grupo experimental que en el grupo control del estudio |
|||
| Tabla 4. Número de fármacos con problemas de seguridad diferentes de los efectos adversos observados clínicamente | |||
| N° a | |||
| Fracasados en el primer ciclo de revisión. (n=151) | Aprobación demorara tras el reenvío (n=71) | Medicamentos nunca aprobados durante el estudio (n=80) | |
| Estudios de seguridad inadecuados o ausentes | |||
| Estudios de prolongación QT | 7 (4,6) | 5 (7,0) | 2 (2,5) |
| Estudios de encimas CYP | 3 (2,0) | 2 (2,8) | 1 (1,3) |
| Estudios de carcinogenicidad | 3(2,0) | 2 (2,8) | 1 (1,3) |
| Estudios de toxicidad reproductiva | 1 (0,7) | 0 | 1 (1,3) |
| Riesgos potenciales basados en toxicología animal (ejemplo: carcinogenicidad) | 8 (5,3) | 4 (5,6) | 4 (5,0) |
| Riesgos teóricos relacionados con el mecanismo de acción o la estructura o clase del fármaco | 11 (7,3) | 7 (9,9) | 4 (5,0) |
| Riesgos potenciales para poblaciones no examinadas | |||
| Población demasiado pequeña para caracterizar la seguridad de fármaco | 2 (1,3) | 1 (1,4) | 1 (1,3) |
| Población de evaluación de la seguridad inadecuada para la dosis/duración de la terapia propuesta | 5 (3,3) | 4 (5,6) | 1 (1,3) |
| Población inadecuada para establecer la seguridad en pacientes con insuficiencia renal /hepática | 7 (4,6) | 4 (5,6) | 3 (3,8) |
| Selección de la dosis | 24 (15,9) | 9 (12,7) | 15 (18,8) |
| Abreviaturas: CYP:: citocromo P a No todas los medicamentos fracasados presentaron problemas de seguridad significativos. Ver Tablas 2,3 y 5 para otras deficiencias |
|||
Veinte medicamentos (13,2%) fracasaron debido a que la inconsistencia en resultados entre múltiples variables objetivos predefinidas en los estudios clínicos impedía la aprobación. Inconsistencia en eficacia para porciones de la población en estudio impidi eron la aprobación de 17 medicamentos (11,3%).
En estos casos, se veía normalmente solo en algunos estudios o en algunos lugares y no en otros o solo en subpoblaciones que no formaban parte del plan analítico inicial.
| Tabla 5. Frecue ncia de deficiencias de seguridad, eficacia, CMC y prospecto en los medicamentos fracasados en el primer ciclo de revisión | |||||
| N° a | |||||
| Fracasados en el primer ciclo de revisión. (n=151) | Aprobación demorara tras el reenvío (n=71) | Medicamentos nunca aprobados durante el estudio (n=80) | P-valor | ||
| Deficiencias solo de eficacia | 48 (31,8) | 15 (21,1) | 33 (41,3) | 0,01 | |
| Deficiencia de eficacia y seguridad | 41 (27,2) | 13 (18,3) | 28 (35,0) | 0,03 | |
| Deficiencia solo de seguridad | 39 (25,8) | 24 (33,8) | 15 (18,8) | 0,04 | |
| CMC solo | 17(11,3) | 13 (18,3) | 4 (5,0) | 0,02 | |
| Prospecto únicamente | 4 (2,6) | 4 (5,6) | 0 | 0,05 | |
| CMC y prospecto | 2 (1,3) | 2 (2,8) | 0 | 0,22 | |
| Abreviaturas: CMC :: química, fabricación y contro | |||||
Hubo 20 medicamentos (13,2%) que a pesar de mostrar superioridad respecto a placebo, se consideró que no tenían suficiente eficacia en comparación con el patrón de cuidado. Los ejemplos incluyen medicamentos dirigidos hacia indicaciones graves (por ejemplo, tratamiento de arritmias, alivio del cáncer, y esquizofrenia) para las cuales ya existen en el mercado productos aprobados más efectivos y los ensayos no mostraron otras ventajas (por ejemplo, mejor seguridad o tratamiento de rescate).
Seguridad. En general, las preocupaciones sobre seguridad, fueron el resultado de los efectos adversos observados en los ensayos clínicos (Tabla 3) que fueron de la suficiente gravedad como para afectar significativamente a la salud del paciente (por ejemplo; accidente cerebrovascular, infarto de miocardio, hepatitis, fracaso renal, ideación suicida y sangrado relacionados con el medicamento).
Para 11 medicamentos que fracasaron en lograr la aprobación en la revisión inicial, las tasas de mortalidad en los ensayos clínicos fueron numéricamente más altas entre los pacientes tratados con el nuevo medicamento que entre los pacientes tratados con el medicamento de comparación. Ninguno fue posteriormente aprobado durante el periodo de estudio. Otras preocupaciones sobre seguridad que impidieron o retrasaron la aprobación del medicamento incluyen estudios clínicos y no clínicos inadecuados o desaparecidos, que usualmente se incluyen en las solicitudes de aprobación de nuevos medicamentos. En varias solicitudes, la población estudiada fue demasiado pequeña o inadecuadas para caracterizar los riesgos anticipados durante el uso clínico. En otros, no se estudiaron riegos teóricos surgidos de señales en estudios animales o de la estructura o mecanismo de acción del fármaco. Preocupaciones sobre la selección de la dosis también se identificaron frecuentemente (Tabla 4).
Prospecto-etiquetado y química-fabricación-control (CMC). Limitamos nuestro análisis sobre deficiencias de etiquetado-prospecto y CMC a los productos que no fueron aprobados a pesar de una eficacia y seguridad satisfactoria (n=23). Las deficiencias de CMC incluyeron problemas con las especificaciones de disolución o fabricación, datos incompletos de estabilidad, niveles elevados de endotoxinas, y deficiencias apreciadas durante la inspección de las fábricas.
La Tabla 5 muestra la frecuencia de deficiencias en seguridad, eficacia, o ambas, y aquellas en el etiquetado-prospecto y de fabricación suficientemente significativas como para evitar la aprobación en ausencia de problemas de eficacia o seguridad. Deficiencias de eficacia fueron evaluadas para 89 medicamentos (48 con deficiencias de eficacia y 41 con deficiencias tanto de eficacia como de seguridad), deficiencias de seguridad se evaluaron en 80 medicamentos (39 con deficiencias de seguridad y 41 con ambas deficiencias de seguridad y eficacia), deficiencias de CMC en 19 y deficiencias en prospecto en 6. La frecuencia de deficiencia de seguridad fue similar entre los medicamentos que nunca se aprobaron en comparación con aquellos con aprobación demorada (43 de 80 nunca aprobados [53,8%] vs 37 de 71 eventualmente aprobados [52,1%]; diferencia 1,7% [IC95%, -14,86% a 18,05%]; p=0,87). Sin embargo, las deficiencias en eficacia fueron significativamente más frecuentes entre los medicamentos nunca aprobados que entre aquellos con aprobación demorada (61 de 80 nunca aprobados 76,3%] vs 28 de 71 eventualmente aprobados [39,4%]; diferencia, 36,9% [CI95%, 20,25% a 50,86%]; p<0,001). Entre los 48 medicamentos con solo preocupaciones de eficacia iniciales, únicamente 31,3% fueron eventualmente aprobadas frente a 61,5% de los 39 medicamentos con solo preocupaciones de seguridad.
Discusión
Entre los años 2000 y 2012, 151 de 302 NMEs (50%) fracasaron en la obtención de autorización de puesta en el mercado, cuando fueron sometidas por primera vez a la FDA. De estas, 71 fueron aprobadas durante subsiguientes sometimientos, con una mediana de 435 días de demora, la tasa global de aprobación fue del 73,5% al final de nuestro estudio. Las solicitudes de aprobación que fueron eventualmente aprobadas fueron a menudo capaces de reconducir problemas iniciales de seguridad, fabricación e información contenida en el prospecto, pero los problemas de eficacia tuvieron menos probabilidad de ser reconducidos con éxito.
Los medicamentos que fracasan durante los estadios finales de su desarrollo, son costosos, a menudo implican el compromiso de muchos participantes y personal de los estudios. Es ventajoso identificar los productos que van a fracasar tan temprano como sea posible a lo largo del proceso de desarrollo para evitar estos problemas. Para aquellos medicamentos que requieren un nuevo sometimiento a revisión antes de ser aprobados, los retrasos están grabando a las industrias y a los reguladores, y los pacientes tienen que esperar para acceder a prometedores, y en ocasiones vitales, nuevos tratamientos [9].
En el momento en que el medicamento entra en sus últimos estadios de su desarrollo, una extensa cantidad de información clínica y no clínica está ya disponible, y los promotores frecuentemente confían en la seguridad y potencial eficacia del fármaco en investigación. Los ensayos en fase 3 proporcionan una oportunidad para caracterizar el tamaño y la naturaleza del efecto clínico y del espectro y frecuencia de las respuestas adversas, permitiendo el desarrollo de detallados e informativos prospectos de los medicamentos. ¿Por qué entonces los medicamentos fracasan en un estado tan avanzado de su desarrollo?
Hemos encontrado que algunos medicamentos inevitablemente fracasan debido a que han probado no ser efectivos o seguros y otros fracasan porque los datos fueron inadecuados para poder evaluar la seguridad y la eficacia.
El fracaso en determinar la dosis más adecuada para el uso clínico fue la principal razón para el rechazo de la aprobación. La dosificación es frecuentemente decidida tempranamente en el desarrollo del fármaco, y la optimización de las dosis para maximizar la eficacia y minimizar la toxicidad es raramente explorada de manera formal en los estudios de fase 3. Diseños adaptativos para los ensayos clínicos [10] y otras estrategias (tales como tratar a los participantes de los ensayos de fase 3 con secuencias aleatorizadas de diferentes dosis [11]) puede ayudar a optimizar las dosis.
Las preocupaciones sobre la eficacia de NMEs fueron una razón frecuente para el fracaso y ha mostrado ser la más difícil de reconducir. Al volver a solicitar la aprobación de comercialización, los medicamentos con preocupaciones sobre la eficacia tuvieron menos probabilidad de alcanzar finalmente la aprobación que aquellos con problemas de seguridad, los cuales pueden ser potencialmente reconducidos con una adecuada información en el prospecto y programas de gestión de riesgos. Hallazgos similares han sido notificados por otros [12,13], y los fracasos tardíos debidos a problemas de eficacia se han asociado con un optimismo prematuro de los promotores relacionado con datos de estudios de fase 2. Solo el 31,3% de los medicamentos con solo problemas de eficacia fueron eventualmente aprobados comparados con el 61,5% de los medicamentos con solo problemas de seguridad.
Entre las solicitudes de comercialización que fracasaron en probar la eficacia, la elección de las variables resultados a medir fueron a menudo inadecuadas para demostrar un beneficio clínicamente significativo para los pacientes [14] (p.e., alivio del dolor, supervivencia, o cura duradera). Imperfectas variables resultado subrogadas se aceptaron en algunas enfermedades (p.e., la prueba de 6 minutos de marcha en hipertensión arterial pulmonar [15]). En otras, como la fibrosis quística [16], la enfermedad de Alzheimer [17] y el cáncer [18], son críticas variables resultado adecuadas a largo plazo para la aprobación y una respuesta temprana puede no trasladarse a respuestas duraderas [19].
Cuando se emplean múltiples variables resultado en un ensayo clínico, y se obtiene resultados discordantes entre ellos, la FDA frecuentemente concluye que la eficacia del medicamento no ha sido probada. Los hallazgos inconsistentes utilizando más de una variable resultado para la misma enfermedad están plagados de variables subrogadas y medidas biológicas que no han sido validadas [20], nosotros hemos encontrado que estas son más frecuentes en medicamentos que nunca fueron aprobados que en aquello los que la aprobación se demoró. La aprobación se negó también cuando la eficacia del nuevo medicamento se juzgó peor que la de los cuidados patrón, de modo que el riesgo sobrepasaba los beneficios. Los investigadores frecuentemente sobreestiman el tratamiento del efecto cuando planifican ensayos clínicos aleatorizados [21,22], y el beneficio clínico del nuevo medicamento puede no ser adecuado para justificar la aprobación, particularmente cuando otros tratamientos están disponibles.
Las preocupaciones sobre la seguridad que más frecuentemente dificultan la aprobación fueron los sucesos clínicos adversos que se producen en los ensayos de fase 3, particularmente aquellos que afectan al sistema cardiovascular. La alta frecuencia de efectos cardiacos adversos puede ser parcialmente debida al amplio abanico de condiciones incluidas en esta categoría (sucesos trombóticos, arritmias, y otras toxicidades cardíacas). Sin embargo, las toxicidades cardíacas pueden escapar a su detección hasta fases tardías del desarrollo del medicamento debido a la pobreza de los métodos predictivos [23]. También, en años recientes, el elevado perfil de retirada por las compañías de inhibidores de la ciclooxigenasa 2 (COX2) han sensibilizado a la comunidad de desarrollo de medicamentos hacia problemas cardiacos similares en otras solicitudes de aprobación de nuevos medicamentos [24] y hay una creciente resistencia a aceptar ciertos niveles de riesgo, en ausencia de una clara necesidad de salud pública insatisfecha. Incluso en programas muy prolongados de desarrollo puede carecer del poder para identificar efectos adversos graves raros. Como parte de la aproximación de ciclo vital en la regulación de medicamentos, la vigilancia de seguridad debe continuar durante todo el tiempo que estos permanezcan en el mercado. Aunque los nuevos hallazgos frecuentemente son incorporados en las actualizaciones del prospecto, es inusual encontrar problemas mayores en productos ya autorizados. De todos los NMEs autorizados durante los 12 años de nuestro estudio, solo 1 fue posteriormente retirado del mercado por problemas de seguridad. Se consideró que valdecoxib tenía un desfavorable cociente riesgo-beneficio como consecuencia de sus graves efectos adversos cardíacos y cutáneos y fue retirado del mercado en 2005 [25].
Altas tasas de aprobación en primer ciclo se han asociado con medicamentos a los que se otorgó una revisión prioritaria para el tratamiento de condiciones graves para los cuales se anticipó mejorías significativas en seguridad o eficacia respecto de las terapias disponibles. En comparación con medicamentos que replicaban el arsenal existente, las que cualificaron para este incentivo se dirigían hacia áreas de necesidades médicas con beneficios que frecuentemente sobrepasaban los riesgos.
Las limitaciones de este estudio incluyen la falta de una métrica objetiva para determinar porque los medicamentos fracasaron en alcanzar la aprobación de comercialización. En ausencia de un método patrón, hemos empleado una aproximación heurística para categorizar las razones por las que no se rechaza la aprobación. En muchos casos la decisión de aprobación fue sencilla. En casos más conflictivos, la FDA generalmente ha confiado en las reuniones del comité asesor con expertos consultores que ayudaron en las decisiones reguladoras. Durante estas reuniones, los datos de las solicitudes de aprobación fueron presentadas por el patrocinador y los revisores de la FDA, y las razones potenciales para el fracaso del medicamento discutidas en profundidad por los miembros del comité [6]. Inevitablemente, las sutilezas de estas complejas decisiones reguladoras no pueden ser capturadas completamente en un análisis agregado.
Conclusiones
Creemos que el juicio de consenso de los experimentados revisores de la FDA sobre las razones para el fracaso de medicamentos ha proporcionado datos descriptivos informativos. La oportunidad para combinar los datos de un gran número de solicitudes de comercialización nos ha permitido identificar categorías de fracaso de medicamentos y su frecuencia relativa a pesar de la incertidumbre asociada con ciertas solicitudes individuales. Nuestros hallazgos pueden ser útiles para los clínicos, y los que toman decisiones, para interpretar la extensa bibliografía que informa sobre el diseño y resultados de ensayos clínicos, los cuales a su vez pueden tener un impacto sobre la práctica. Para los que desarrollan los fármacos y los investigadores clínicos, nuestros hallazgos sugieren áreas de deficiencias en las solicitudes de comercialización de nuevos medicamentos para las cuales pueden desarrollarse estrategias de mejora durante el desarrollo de nuevos fármacos. El dialogo temprano y frecuente entre la FDA y los patrocinadores para conducir los aspectos críticos del diseño de los estudios (incluyendo la selección de la población de estudio, variables resultado del estudio, y dosis del medicamento) tiene un potencial para reducir el retraso en la aprobación de nuevos fármacos [26].
Información sobre el artículo: los autores declararon no tener conflicto de intereses. Las opiniones vertidas en el artículo corresponden a los autores y no reflejan necesariamente las opiniones oficiales de la FDA
Referencias