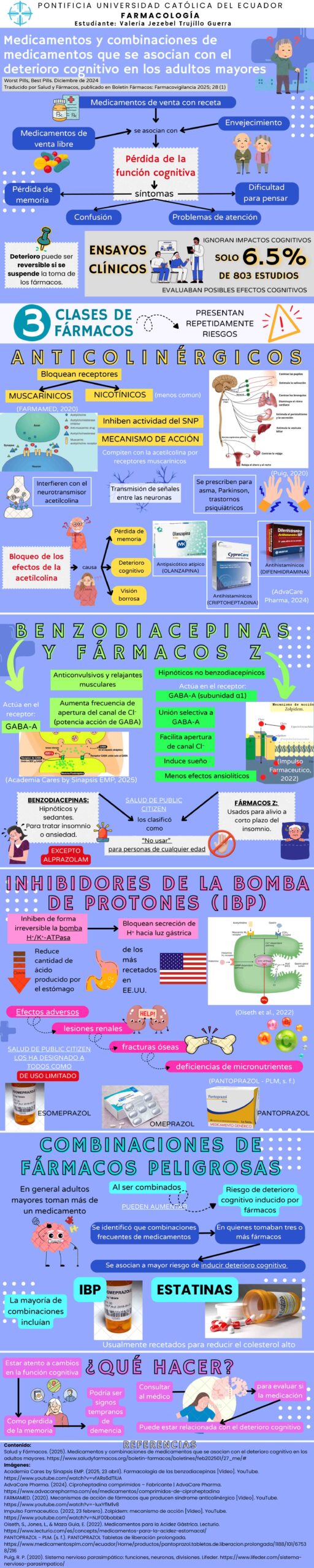
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Reflexiones alrededor de las muertes relacionadas con terapias génicas para la Distrofia Muscular
Natalia Castrillón Valencia MD
ConsultorSalud, 28 de julio de 2025
https://consultorsalud.com/reflexiones-terapias-genicas-distrofia-muscular/
Tags: muertes relacionadas con terapias génicas para la Distrofia Muscular, efectos adversos graves de terapia génica para tratar Distrofia Muscular, fallecimientos por insuficiencia hepática aguda vinculados a terapias génicas de Sarepta, muertes relacionadas con Elevidys, muerte en ensayo clínico SRP-9004
Sarepta Therapeutics enfrenta una creciente crisis de confianza tras la notificación de tres muertes asociadas a sus terapias génicas, dos casos vinculados a Elevidys, el tratamiento para la Distrofia Muscular de Duchenne (DMD, por su sigla en inglés Duchenne Muscular Dystrophy) y un caso asociado a la terapia experimental para la Distrofia Muscular de Cinturas (LGMD, por su sigla en inglés Limb Girdle Muscular Dystrophy).
La DMD es una enfermedad hereditaria ligada al cromosoma X y es la distrofia muscular infantil más común (afecta a 1 de cada 5.000 varones) [1]. Se caracteriza por la atrofia muscular progresiva, con pérdida de la capacidad para caminar, dependencia total de la silla de ruedas hacia los 12 o 13 años, y muerte en la tercera década de la vida por complicaciones respiratorias y cardíacas. Desarrollar una terapia eficaz para frenar el deterioro muscular y prolongar la capacidad de caminar y la expectativa de vida continúan siendo objetivos terapéuticos prioritarios [2].
La FDA otorgó la aprobación acelerada a Elevidys en 2023 y se ha administrado cientos de pacientes. En junio de 2024 se produjo la primera muerte de un adolescente de 16 años que había recibido dicho tratamiento y desarrolló insuficiencia hepática [3], reactivando las alertas sobre la seguridad de la terapia.
Aunque Sarepta atribuyó el desenlace fatal a una posible coinfección por citomegalovirus, la limitada evidencia clínica presentada para su aprobación (incluyendo solo 16 pacientes mayores de 8 años y 8 no ambulatorios), cuestionó la validez externa y la representatividad del ensayo clínico pivotal, sin desconocer el contexto de las enfermedades huérfanas.
La muerte de un segundo adolescente por insuficiencia hepática tras recibir la terapia génica Elevidys, ha generado temor y división en la comunidad de pacientes con DMD. En este caso, los síntomas relacionados con insuficiencia hepática empezaron a aparecer seis semanas después de haber iniciado la terapia génica [4].
Las Distrofias Musculares de Cinturas (LGMD) son trastornos hereditarios heterogéneos sin cura, que incluyen 29 formas recesivas (LGMDR) y 5 formas dominantes (LGMDD) [5], con inicio en la infancia o adultez temprana y caracterizadas por debilidad muscular proximal progresiva, variabilidad fenotípica, que a veces cursa con afectación cardíaca o respiratoria y a medida que va progresando genera dependencia de caminador o silla de ruedas.
Encontrar una cura para la LGMD es difícil debido a su lenta evolución, por lo que la recopilación exhaustiva de datos a través de registros, redes y estudios de su historia natural es fundamental. Los estudios y registros son limitados y dispersos, dificultando su estandarización y el seguimiento [5].
Sarepta, en una carta dirigida a la comunidad con DMD el pasado 19 de julio [6] menciona que el tercer caso de muerte ocurrió en un ensayo clínico fase 1 (SRP-9004) con una terapia génica experimental diferente para tratar LGMDR tipo 2D. La compañía asegura que reportó ese fallecimiento a la FDA el 3 de julio y que el 20 de junio había notificado al ente regulador que la condición clínica del participante en el ensayo SRP-9004 se estaba tornando potencialmente fatal (un paciente adulto que murió por falla hepática). Según Whitlock [7], la compañía no divulgó este evento en su comunicación a inversionistas, lo que motivó críticas por parte de analistas y generó una pérdida bursátil del 27%.
Estos eventos adversos fatales, junto con los cuestionamientos sobre el proceder corporativo y el programa regulatorio de aprobación acelerada [8] han intensificado el debate sobre la seguridad de las terapias génicas, particularmente en poblaciones pediátricas y en enfermedades huérfanas.
La EMA, aunque también cuenta con políticas de aprobación condicional para enfermedades graves sin alternativas terapéuticas [9], mantiene criterios más estrictos para la demostración de beneficio clínico y planes de seguimiento a largo plazo que los que la FDA aplicó en el caso de Elevidys.
La vía acelerada de la FDA, diseñada para facilitar el acceso temprano a terapias prometedoras para condiciones graves, ha sido objeto de cuestionamientos por permitir que se tomen decisiones basadas en criterios de valoración indirectos o subrogados y en poblaciones no representativas.
Utilizar datos subrepresentativos debilita la calidad del consentimiento informado, pues los riesgos reales no se cuantifican adecuadamente y por ende el consentimiento se otorga a pesar de los vacíos de conocimiento.
La aprobación acelerada de Elevidys, favorecida por funcionarios que ignoraron las objeciones técnicas de los revisores internos [10], sugiere la presencia de un riesgo regulatorio o de presión externa que socava la independencia técnica del proceso. La EMA, en contraste, exige planes detallados de seguimiento y reevaluación, aunque también ha enfrentado críticas similares en otras ocasiones.
Se suman tres fallecimientos vinculados a las terapias génicas de Sarepta, todos por presunta insuficiencia hepática aguda (Ver Cuadro 1), reactivando la controversia que rodeó la aprobación acelerada de Elevidys.
Estos casos también generan dilemas clínicos y éticos por su afectación a poblaciones pediátricas y adultos frágiles, especialmente vulnerables y sin acceso a otros tratamientos efectivos.
Se debería estudiar si los eventos descritos se hubieran podido prevenir con una mejor adherencia a los principios fundamentales de la bioética, particularmente los establecidos por las Pautas Éticas Internacionales del CIOMS y la declaración de Helsinki.
La Pauta 15 de CIOMS [11] enfatiza que toda investigación con personas en situaciones de vulnerabilidad debe tener una justificación científica sólida y debe al mismo tiempo minimizar el riesgo mediante evidencia preclínica robusta y criterios de inclusión claros; la Pauta 23 resalta que la transparencia de las comunidades científicas y los entes reguladores es esencial para mantener la confianza pública.
Es conocido que el uso de terapias génicas implica riesgos inherentes, en especial los relacionados con hepatotoxicidad, como se ha documentado también con onasemnogene abeparvovec (Zolgensma) [12], que se utiliza para tratar la atrofia muscular espinal.
El numeral 16 de la declaración de Helsinki [13] enfatiza que los beneficios de la investigación deben superar los riesgos potenciales para los participantes, y el numeral 18 subraya la importancia de que los médicos evalúen cuidadosamente los riesgos y los beneficios potenciales y se aseguren de que los riesgos pueden ser manejados de manera satisfactoria antes de iniciar la investigación. En este caso, la relación riesgo/beneficio de las terapias génicas para los afectados podría haber sido sobrestimada.
Estos acontecimientos recientes relacionados con terapias génicas para la Distrofia Muscular revelan un desequilibrio preocupante entre la necesidad de innovación tecnológica en salud y la obligación ética de proteger la vida de los participantes en ensayos clínicos y de los usuarios de las tecnologías aprobadas.
El desarrollo de terapias génicas representa un avance de la ciencia en la búsqueda de tratamientos para enfermedades donde históricamente solo había cuidados paliativos. Sin embargo, conforme a las Pautas Éticas Internacionales [11, 13] es imperativo subrayar que la innovación biomédica no puede desligarse del deber de proteger la vida y la seguridad de quienes acuden a la esperanza de la experimentación confiando en la idoneidad científica y regulatoria.
La muerte de los dos adolescentes impacta profundamente a la comunidad con DMD, que enfrenta una enfermedad progresiva y letal, y que ahora, además, carga con la incertidumbre de elegir entre la expectativa de vida esperada con los cuidados convencionales o los riesgos de una terapia novedosa (para quienes esta opción es asequible).
Una reflexión más profunda debería abarcar las dudas éticas sobre la proporcionalidad del riesgo, la calidad del consentimiento informado, la revisión crítica de los procesos de desarrollo, evaluación y aprobación de tecnologías en salud, la asequibilidad de terapias de alto costo y el equilibrio entre la urgencia por acceder a tratamientos innovadores frente a la flexibilización de los estándares éticos, metodológicos y regulatorios.
Referencias:
La FDA advierte sobre el riesgo grave de complicaciones por calor asociado al parche antinaúseas transdérmico de escopolamina (Transderm Scōp)
(FDA issues warning about serious risk of heat-related complications with the antinausea patch scopolamine transdermal system transderm scōp)
Worst Pills Best Pills, 14 de julio de 2025.
https://www.worstpills.org/e-alerts/view/147
Traducido por Salud y Fármacos
Tags: grave riesgo de complicaciones relacionadas con transderm scōp, el parche antinaúseas transderm scōp puede causar graves complicaciones, muerte en niños y adultos mayores asociada al uso de transderm scōp
El 18 de junio de 2025, la FDA emitió una advertencia de seguridad para quienes usan el sistema transdérmico de escopolamina (transderm scop), un parche que se utiliza para tratar las náuseas y los vómitos, pues pueden experimentar complicaciones relacionadas con el calor [1]. El parche puede aumentar la temperatura corporal y disminuir la sudoración, lo que puede provocar complicaciones relacionadas con el calor, como confusión, pérdida del conocimiento, hospitalización o, en algunos casos, incluso la muerte.
La FDA recomienda que cualquier persona, especialmente los menores de 17 años y los adultos mayores de 60 años, que experimente síntomas de hipertermia como temperatura corporal elevada o disminución de la sudoración se retire el parche inmediatamente. Estas personas también deben evitar el uso de fuentes de calor externas como mantas térmicas, y deben mantenerse alejados de ambientes cálidos.
El sistema transdérmico de escopolamina, aprobado inicialmente en 1979, es un parche que se utiliza en adultos para tratar las náuseas y los vómitos asociados con el mareo por movimiento y la recuperación de la anestesia [2]. El parche para tratar las náuseas funciona liberando escopolamina, un anticolinérgico que bloquea las señales cerebrales responsables de las náuseas y los vómitos. El parche se aplica detrás de la oreja y libera un miligramo de escopolamina, cuyo efecto antiemético (evitar el vómito), comienza en cuatro horas y dura hasta tres días.
Las personas con glaucoma de ángulo cerrado o hipersensibilidad a la escopolamina no deben utilizar el parche. Aunque no está aprobado para su uso en niños, el sistema transdérmico de escopolamina se ha recetado ocasionalmente fuera de indicación para tratar la salivación excesiva en niños con parálisis cerebral u otros trastornos neurológicos [3].
La advertencia de seguridad sobre la escopolamina transdérmica se ha actualizado para añadir el riesgo de hipertermia a la información para la prescripción y al prospecto para el paciente. Los agentes anticolinérgicos como la escopolamina pueden elevar la temperatura corporal central y disminuir la sudoración. Estos efectos pueden agravarse por la exposición a fuentes externas de calor o en entornos con altas temperaturas. Estos síntomas, si no se detectan o no se tratan, pueden provocar complicaciones graves. En la mayoría de los casos la hipertermia se presenta dentro de las 72 horas posteriores a la aplicación. Incluso después de retirar el parche, los síntomas de abstinencia como mareos, dolor de cabeza y náuseas, pueden persistir durante varios días, ya que el medicamento absorbido permanece en el organismo [4].
La advertencia de la FDA se basa en 13 casos de hipertermia reportados a nivel global en pacientes que usan el parche para tratar las náuseas, incluyendo siete en EE UU. De estos casos, ocho involucraron a niños menores de 17 años y cuatro a adultos mayores de 60 años. Cuatro casos resultaron en hospitalización y dos pacientes murieron: uno era un niño y el otro un adulto mayor.
“En base al funcionamiento del medicamento, incluyendo su capacidad para atravesar la barrera hematoencefálica, una membrana que impide que sustancias potencialmente dañinas de la sangre lleguen al cerebro, la FDA determinó que existe evidencia razonable de asociación causal entre los parches de escopolamina y la hipertermia” [4].
El Grupo de Investigación en Salud de Public Citizen recomienda que, si utiliza el sistema transdérmico de escopolamina, esté atento a cualquier signo de calor excesivo o alteración de la temperatura, y que si presenta síntomas se retire el parche inmediatamente y se ponga en contacto con su médico.
Para consultar la alerta de la FDA, visite el siguiente enlace: https://www.fda.gov/media/187121/download
Informe cualquier evento adverso asociado con la escopolamina transdérmica (Transderm Scōp) al programa MedWatch de la FDA llamando al 1-888-463-6332 o visitando https://www.accessdata.fda.gov/scripts/medwatch/
Referencias
Petro del lado de la industria farmacéutica transnacional
Bernardo Useche Aldana
La Tribuna, 13 de julio 2025
https://latribunacolombia.co/petro-del-lado-de-la-industria-farmaceutica-transnacional/
Tags: Acceso universal a medicamentos, justicia sanitaria global, reformar sistema de patentes, Propiedad intelectual y salud pública, Industria farmacéutica transnacional, Comités de ética en investigación, Ensayos clínicos en América Latina.
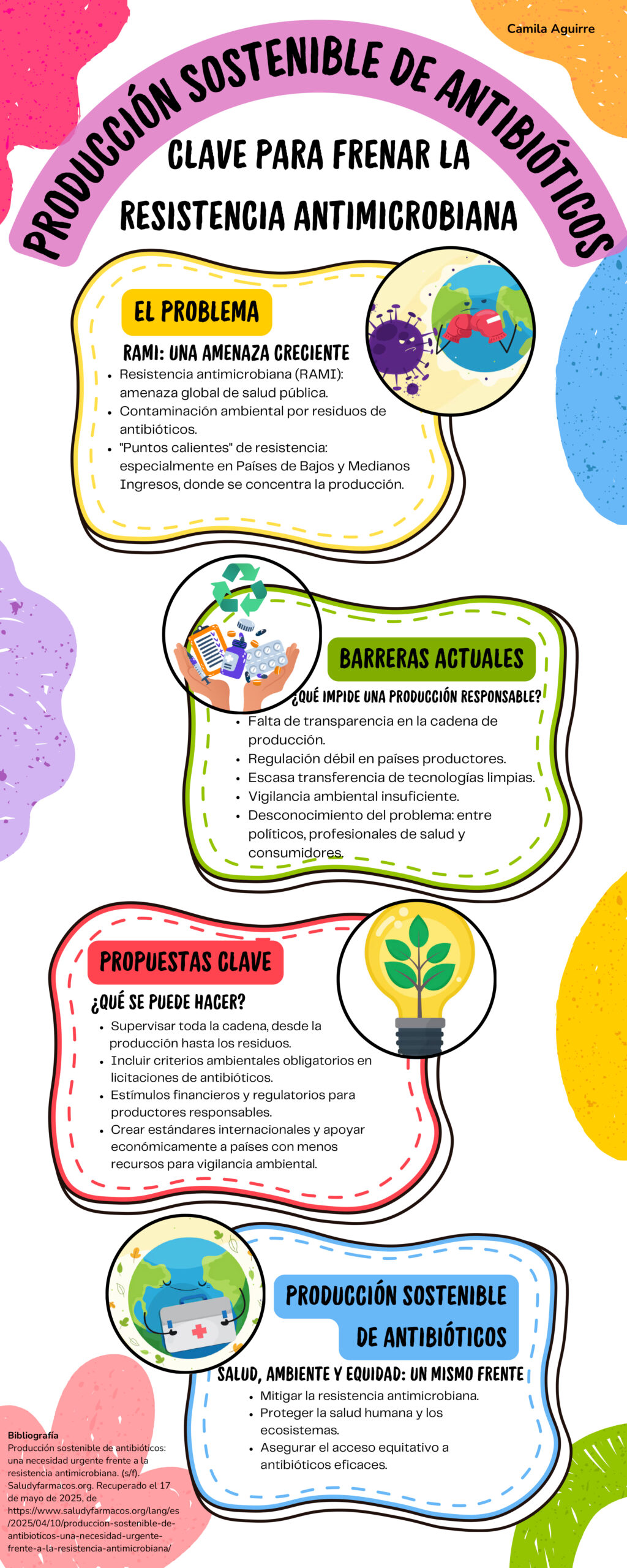
La industria farmacéutica transnacional, con lineamientos del Banco Mundial, impulsa un plan para convertir a Colombia en centro regional de ensayos clínicos en América Latina. Declaraciones empresariales muestran que se busca redirigir recursos públicos, reformar los comités de ética y ampliar la presencia de multinacionales en el sistema de salud.
La Ley 100 introdujo como actores principales en el sistema a las EPS y el modelo de aseguramiento social en salud. En el panorama que se avecina luego del desbarajuste ocasionado con la política implementada por el presidente Petro y sus ministros de salud, las multinacionales farmacéuticas representadas por la Asociación de Laboratorios Farmacéuticos de Investigación y Desarrollo, AFIDRO, buscan jugar un rol preponderante en el nuevo sistema de salud que termine por configurarse.
El objetivo va más allá: “consolidar a Colombia como un hub regional de investigación” en América Latina. El interés de la industria farmacéutica en este empeño se basa en el hecho de que el mercado farmacéutico latinoamericano es uno de los de mayor crecimiento en el mundo con una población creciente de 660 millones adultos mayores. Interés incentivado por las recientes órdenes ejecutivas expedidas por el presidente Trump con el propósito de acelerar la producción de nuevas medicinas y disminuir los precios de los medicamentos en Estados Unidos, lo que obliga a las grandes farmacéuticas a aumentar los precios y los mercados en el exterior (Executive Order 14273 of April 15, 2025; Executive Order 14293 of May 5, 2025 – Executive Order 14297 of May 12, 2025).
El papel del Banco Mundial
Si en 1993 la reforma neoliberal de salud en Colombia en beneficio de las compañías aseguradoras en salud fue diseñada por el Banco Mundial, hoy, el Banco ha definido también las líneas centrales de la política de participación de las gran (https://openknowledge.worldbank.org/entities/publication/e5fae256-f0c0-5400-9c14-b80387e182c8) industria farmacéutica en los sistemas de salud en América Latina y el Caribe:
Las orientaciones del Banco Mundial se han materializado en una estrategia formulada por la Federación Latinoamericana de la Industria Farmacéutica (https://afidro.org/wp-content/uploads/2025/06/FIFARMA-Investigacion_Clinica_en_Latinoamerica_2.pdf), organización que agrupa “empresas farmacéuticas multinacionales líderes” y que actualmente está bajo la dirección de Yaneth Giha, exdirectora de AFIDRO y Ministra de Educación en el segundo gobierno de Juan Manuel Santos.
Aseguradoras y farmacéuticas
El propósito de los grandes laboratorios y compañías farmacéuticas es en primer lugar, articularse a los sistemas de salud y elevar las ventas de sus medicamentos y tecnologías con cargo a los recursos del Estado. Otro documento de la federación es claro al respecto: “El llamado que hacemos desde FIFARMA (…) es que los gobiernos de la región pongan en marcha planes de incremento en sus presupuestos para el sector salud”.
Aseguradoras y farmacéuticas coinciden en que se aumente el presupuesto general en salud, pero tienen entre sí una contradicción económica. A mayor gasto en medicamentos menor presupuesto disponible para el aseguramiento. En la superficie se clama por el aumento del financiamiento público de la salud y de la UPC para responder a las tutelas, quejas y reclamos de la población, lo cual tiene sentido en la crisis actual de la salud, profundizada en el presente gobierno.
En el fondo, las casas farmacéuticas se aprestan a competir con las aseguradoras por un mayor monto de los recursos del Estado. Esto implica una disputa por los servicios y tecnologías que se incluyan en el plan de beneficios y que se contraten con hospitales y clínicas. A propósito, un riguroso estudio reciente del sistema de salud de Taiwán (https://www.sciencedirect.com/science/article/pii/S0929664624004790?via%3Dihub) encontró que a medida que, con recursos del Estado, aumentaron las ganancias de las farmacéuticas, más se afectó la estabilidad financiera de los hospitales.
Más ensayos clínicos en Latinoamérica, parte de la estrategia global de frenar a China
En este contexto, las multinacionales farmacéuticas han lanzado un plan para convertir a Colombia en tierra fértil que produzca un “crecimiento exponencial” de los ensayos clínicos con los que la industria crea productos farmacéuticos de “innovación” cuyas patentes garantizan altísimos precios y monopolios en el mercado.
No extraña entonces que el Dr. Ignacio Gaitán, presidente actual de AFIDRO, haya declarado que: “América Latina podría cuadruplicar su participación en estudios clínicos”. Este plan de expansión de los estudios clínicos en los que se evalúa si un nuevo medicamento tiene efecto preventivo o curativo (eficacia), y si no produce efectos adversos de consideración para la salud de los participantes (seguridad), se encuadra en el reconocimiento por parte de la industria de que “la investigación clínica es una actividad altamente competitiva, que se desarrolla en un entorno global”.
En particular, para las multinacionales norteamericanas incrementar el número de ensayos clínicos en Latinoamérica se inscribe en la estrategia global de la industria de frenar el avance de China en la Investigación y Desarrollo (I & D) de productos farmacéuticos innovadores.
En el período 2000-2004, el número de ensayos clínicos iniciados en China (274) fue 40 veces menor que los estudios emprendidos por Estados Unidos (11.830). En el período 2020-2024, Estados Unidos inició 48.192 ensayos clínicos, solo dos veces el número de los estudios iniciados por China (23.137). De continuar su ritmo de crecimiento, China será en corto tiempo el país a la vanguardia en la investigación farmacéutica, lo que tendrá un gran impacto en el mercado global a mediano plazo.
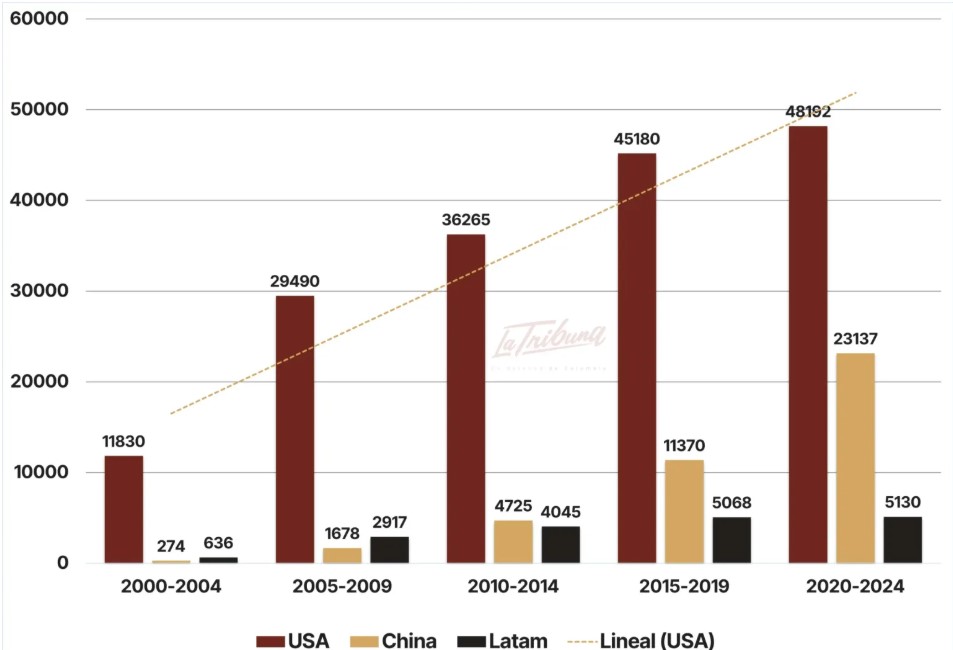
Las grandes farmacéuticas consideran que Colombia y otros países latinoamericanos ofrecen condiciones muy favorables para su política de competir con China en la investigación de nuevos productos. El gasto público en salud crece, la población es étnica y racialmente diversa, el sistema de salud cubre la casi totalidad de la población, los costos de los ensayos clínicos son muy inferiores a los realizados en Europa o Norteamérica.
El gobierno de Gustavo Petro ha visto con buenos ojos el posicionamiento de las multinacionales en el país. Un documento del Ministerio de Salud de 2023 lo plantea: “En Colombia, la investigación clínica patrocinada por la industria farmacéutica ha ido avanzando en los últimos 30 años, posicionándose en el cuarto lugar en Latinoamérica y siendo uno de los países de gran interés por parte de la industria gracias a la infraestructura, calidad de los profesionales de la salud y cobertura del sistema de salud”.
En consonancia con el Ministerio, el mes de mayo pasado, María Fernanda Velasco, directora médica de Pfizer Colombia informó: “Pfizer ha invertido más de 524.5 millones de dólares en investigación clínica desde 2020, posicionando la región como un punto estratégico para la validación científica de tratamientos innovadores para enfermedades complejas que afectan a muchas personas”. La Doctora Velasco agregó que Pfizer cuenta con más de 300 centros de investigación en Brasil y más de 250 en otros países de la región, incluyendo a Colombia.
El papel de los Comités de Ética en Investigación
Ahora, multiplicar de manera expedita los ensayos clínicos requiere que los estudios obtengan rápida aprobación por Comités de Ética de la Investigación (CEI). Estos comités surgieron luego de que una comisión constatara los delitos atroces cometidos durante experimentos médicos con prisioneros en campos de concentración Nazis y otros casos notorios de abuso (Ver Tabla 1).
Posteriormente, en distintos momentos de la historia se ha documentado la existencia de condenables abusos con personas incluidas en diversos estudios clínicos (ver Tabla 2). Se hizo entonces necesario formular criterios y normas éticas y crear los CEI para garantizar la protección de las personas o pacientes participantes en este tipo de estudios.
Las compañías transnacionales de productos farmacéuticos consideran que el funcionamiento de los CEI es lento, que los criterios y normas éticas reguladoras de la investigación clínica se deben unificar internacionalmente y en últimas, que la aprobación de los ensayos debería hacerse en el país “por parte de un único comité”.
La propuesta se orienta a que los CEI sean reemplazados por Organizaciones de Investigación por Contrato o CRO por sus iniciales en inglés (Contract Research Organizations), compañías privadas afines a los intereses de la gran industria farmacéutica.
Los CEI no están exentos de problemas. Un estudio en ocho países latinoamericanos que hicimos bajo la dirección de Nuria Homedes y Antonio Ugalde encontró que, tanto en Colombia como en la región, los comités adolecen de falencias que debilitan su función protectora de los participantes, están expuestos a conflictos de interés con los patrocinadores y tienden a no dar prioridad a proyectos de investigación dirigidos a resolver las necesidades de salud de las poblaciones locales.
Sin embargo, un fuerte sistema de comités de ética independientes de la industria, centrado en la protección de los pacientes y en un conocimiento científico que ofrezca garantía de la seguridad y eficacia de los medicamentos investigados, podría dar mejores resultados que comités con protocolos orientados a favorecer la inversión de las multinacionales.
El dilema de Petro: del lado de las transnacionales o de la industria farmacéutica nacional
El gobierno a través del INVIMA convocó a la Primera Mesa Nacional de Investigación Clínica el pasado 11 de junio donde AFIDRO compartió su política de dar pasos adelante en posicionar al país como un hub en América Latina para el desarrollo de medicamentos que puedan ser patentados por las multinacionales farmacéuticas.
La gran industria farmacéutica debe mantener un lugar importante en el sistema de salud colombiano, pero la industria farmacéutica nacional debe fortalecerse. Las decisiones que se tomen sobre los CEI y las condiciones en que agilicen los trámites del INVIMA y la regularización de los ensayos clínicos van a definir el nivel de incidencia de las multinacionales en el sistema de salud colombiano.
Es una prioridad que se revise el capítulo 16 del TLC con Estados Unidos sobre propiedad intelectual. Es fundamental que la investigación clínica con recursos públicos conduzca a productos con patentes nacionales. Es el único camino para que Colombia se ubique en la senda de la seguridad y la soberanía farmacéutica.