Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Reflexiones alrededor de las muertes relacionadas con terapias génicas para la Distrofia Muscular
Natalia Castrillón Valencia MD
ConsultorSalud, 28 de julio de 2025
https://consultorsalud.com/reflexiones-terapias-genicas-distrofia-muscular/
Tags: muertes relacionadas con terapias génicas para la Distrofia Muscular, efectos adversos graves de terapia génica para tratar Distrofia Muscular, fallecimientos por insuficiencia hepática aguda vinculados a terapias génicas de Sarepta, muertes relacionadas con Elevidys, muerte en ensayo clínico SRP-9004
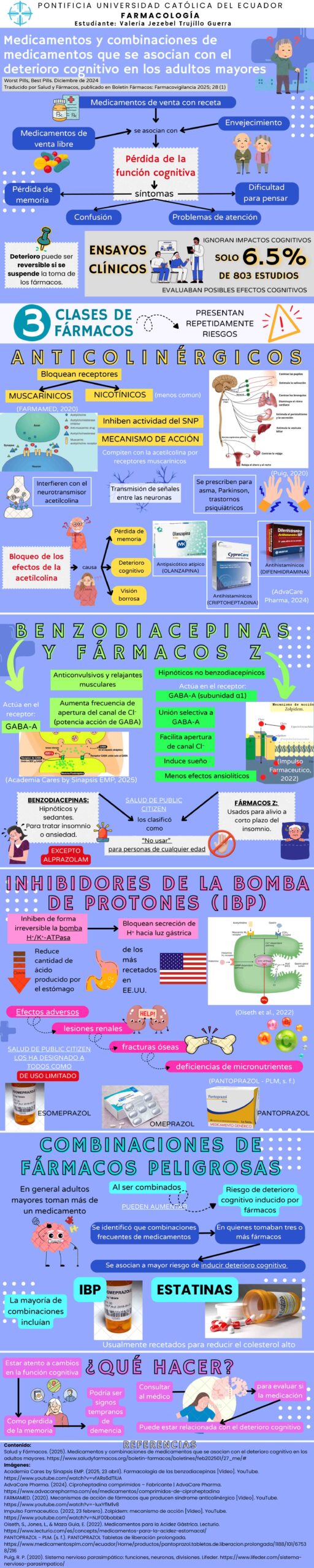
Sarepta Therapeutics enfrenta una creciente crisis de confianza tras la notificación de tres muertes asociadas a sus terapias génicas, dos casos vinculados a Elevidys, el tratamiento para la Distrofia Muscular de Duchenne (DMD, por su sigla en inglés Duchenne Muscular Dystrophy) y un caso asociado a la terapia experimental para la Distrofia Muscular de Cinturas (LGMD, por su sigla en inglés Limb Girdle Muscular Dystrophy).
La DMD es una enfermedad hereditaria ligada al cromosoma X y es la distrofia muscular infantil más común (afecta a 1 de cada 5.000 varones) [1]. Se caracteriza por la atrofia muscular progresiva, con pérdida de la capacidad para caminar, dependencia total de la silla de ruedas hacia los 12 o 13 años, y muerte en la tercera década de la vida por complicaciones respiratorias y cardíacas. Desarrollar una terapia eficaz para frenar el deterioro muscular y prolongar la capacidad de caminar y la expectativa de vida continúan siendo objetivos terapéuticos prioritarios [2].
La FDA otorgó la aprobación acelerada a Elevidys en 2023 y se ha administrado cientos de pacientes. En junio de 2024 se produjo la primera muerte de un adolescente de 16 años que había recibido dicho tratamiento y desarrolló insuficiencia hepática [3], reactivando las alertas sobre la seguridad de la terapia.
Aunque Sarepta atribuyó el desenlace fatal a una posible coinfección por citomegalovirus, la limitada evidencia clínica presentada para su aprobación (incluyendo solo 16 pacientes mayores de 8 años y 8 no ambulatorios), cuestionó la validez externa y la representatividad del ensayo clínico pivotal, sin desconocer el contexto de las enfermedades huérfanas.
La muerte de un segundo adolescente por insuficiencia hepática tras recibir la terapia génica Elevidys, ha generado temor y división en la comunidad de pacientes con DMD. En este caso, los síntomas relacionados con insuficiencia hepática empezaron a aparecer seis semanas después de haber iniciado la terapia génica [4].
Las Distrofias Musculares de Cinturas (LGMD) son trastornos hereditarios heterogéneos sin cura, que incluyen 29 formas recesivas (LGMDR) y 5 formas dominantes (LGMDD) [5], con inicio en la infancia o adultez temprana y caracterizadas por debilidad muscular proximal progresiva, variabilidad fenotípica, que a veces cursa con afectación cardíaca o respiratoria y a medida que va progresando genera dependencia de caminador o silla de ruedas.
Encontrar una cura para la LGMD es difícil debido a su lenta evolución, por lo que la recopilación exhaustiva de datos a través de registros, redes y estudios de su historia natural es fundamental. Los estudios y registros son limitados y dispersos, dificultando su estandarización y el seguimiento [5].
Sarepta, en una carta dirigida a la comunidad con DMD el pasado 19 de julio [6] menciona que el tercer caso de muerte ocurrió en un ensayo clínico fase 1 (SRP-9004) con una terapia génica experimental diferente para tratar LGMDR tipo 2D. La compañía asegura que reportó ese fallecimiento a la FDA el 3 de julio y que el 20 de junio había notificado al ente regulador que la condición clínica del participante en el ensayo SRP-9004 se estaba tornando potencialmente fatal (un paciente adulto que murió por falla hepática). Según Whitlock [7], la compañía no divulgó este evento en su comunicación a inversionistas, lo que motivó críticas por parte de analistas y generó una pérdida bursátil del 27%.
Estos eventos adversos fatales, junto con los cuestionamientos sobre el proceder corporativo y el programa regulatorio de aprobación acelerada [8] han intensificado el debate sobre la seguridad de las terapias génicas, particularmente en poblaciones pediátricas y en enfermedades huérfanas.
La EMA, aunque también cuenta con políticas de aprobación condicional para enfermedades graves sin alternativas terapéuticas [9], mantiene criterios más estrictos para la demostración de beneficio clínico y planes de seguimiento a largo plazo que los que la FDA aplicó en el caso de Elevidys.
La vía acelerada de la FDA, diseñada para facilitar el acceso temprano a terapias prometedoras para condiciones graves, ha sido objeto de cuestionamientos por permitir que se tomen decisiones basadas en criterios de valoración indirectos o subrogados y en poblaciones no representativas.
Utilizar datos subrepresentativos debilita la calidad del consentimiento informado, pues los riesgos reales no se cuantifican adecuadamente y por ende el consentimiento se otorga a pesar de los vacíos de conocimiento.
La aprobación acelerada de Elevidys, favorecida por funcionarios que ignoraron las objeciones técnicas de los revisores internos [10], sugiere la presencia de un riesgo regulatorio o de presión externa que socava la independencia técnica del proceso. La EMA, en contraste, exige planes detallados de seguimiento y reevaluación, aunque también ha enfrentado críticas similares en otras ocasiones.
Se suman tres fallecimientos vinculados a las terapias génicas de Sarepta, todos por presunta insuficiencia hepática aguda (Ver Cuadro 1), reactivando la controversia que rodeó la aprobación acelerada de Elevidys.
Estos casos también generan dilemas clínicos y éticos por su afectación a poblaciones pediátricas y adultos frágiles, especialmente vulnerables y sin acceso a otros tratamientos efectivos.
Se debería estudiar si los eventos descritos se hubieran podido prevenir con una mejor adherencia a los principios fundamentales de la bioética, particularmente los establecidos por las Pautas Éticas Internacionales del CIOMS y la declaración de Helsinki.
La Pauta 15 de CIOMS [11] enfatiza que toda investigación con personas en situaciones de vulnerabilidad debe tener una justificación científica sólida y debe al mismo tiempo minimizar el riesgo mediante evidencia preclínica robusta y criterios de inclusión claros; la Pauta 23 resalta que la transparencia de las comunidades científicas y los entes reguladores es esencial para mantener la confianza pública.
Es conocido que el uso de terapias génicas implica riesgos inherentes, en especial los relacionados con hepatotoxicidad, como se ha documentado también con onasemnogene abeparvovec (Zolgensma) [12], que se utiliza para tratar la atrofia muscular espinal.
El numeral 16 de la declaración de Helsinki [13] enfatiza que los beneficios de la investigación deben superar los riesgos potenciales para los participantes, y el numeral 18 subraya la importancia de que los médicos evalúen cuidadosamente los riesgos y los beneficios potenciales y se aseguren de que los riesgos pueden ser manejados de manera satisfactoria antes de iniciar la investigación. En este caso, la relación riesgo/beneficio de las terapias génicas para los afectados podría haber sido sobrestimada.
Estos acontecimientos recientes relacionados con terapias génicas para la Distrofia Muscular revelan un desequilibrio preocupante entre la necesidad de innovación tecnológica en salud y la obligación ética de proteger la vida de los participantes en ensayos clínicos y de los usuarios de las tecnologías aprobadas.
El desarrollo de terapias génicas representa un avance de la ciencia en la búsqueda de tratamientos para enfermedades donde históricamente solo había cuidados paliativos. Sin embargo, conforme a las Pautas Éticas Internacionales [11, 13] es imperativo subrayar que la innovación biomédica no puede desligarse del deber de proteger la vida y la seguridad de quienes acuden a la esperanza de la experimentación confiando en la idoneidad científica y regulatoria.
La muerte de los dos adolescentes impacta profundamente a la comunidad con DMD, que enfrenta una enfermedad progresiva y letal, y que ahora, además, carga con la incertidumbre de elegir entre la expectativa de vida esperada con los cuidados convencionales o los riesgos de una terapia novedosa (para quienes esta opción es asequible).
Una reflexión más profunda debería abarcar las dudas éticas sobre la proporcionalidad del riesgo, la calidad del consentimiento informado, la revisión crítica de los procesos de desarrollo, evaluación y aprobación de tecnologías en salud, la asequibilidad de terapias de alto costo y el equilibrio entre la urgencia por acceder a tratamientos innovadores frente a la flexibilización de los estándares éticos, metodológicos y regulatorios.
Referencias:
La FDA advierte sobre el riesgo grave de complicaciones por calor asociado al parche antinaúseas transdérmico de escopolamina (Transderm Scōp)
(FDA issues warning about serious risk of heat-related complications with the antinausea patch scopolamine transdermal system transderm scōp)
Worst Pills Best Pills, 14 de julio de 2025.
https://www.worstpills.org/e-alerts/view/147
Traducido por Salud y Fármacos
Tags: grave riesgo de complicaciones relacionadas con transderm scōp, el parche antinaúseas transderm scōp puede causar graves complicaciones, muerte en niños y adultos mayores asociada al uso de transderm scōp
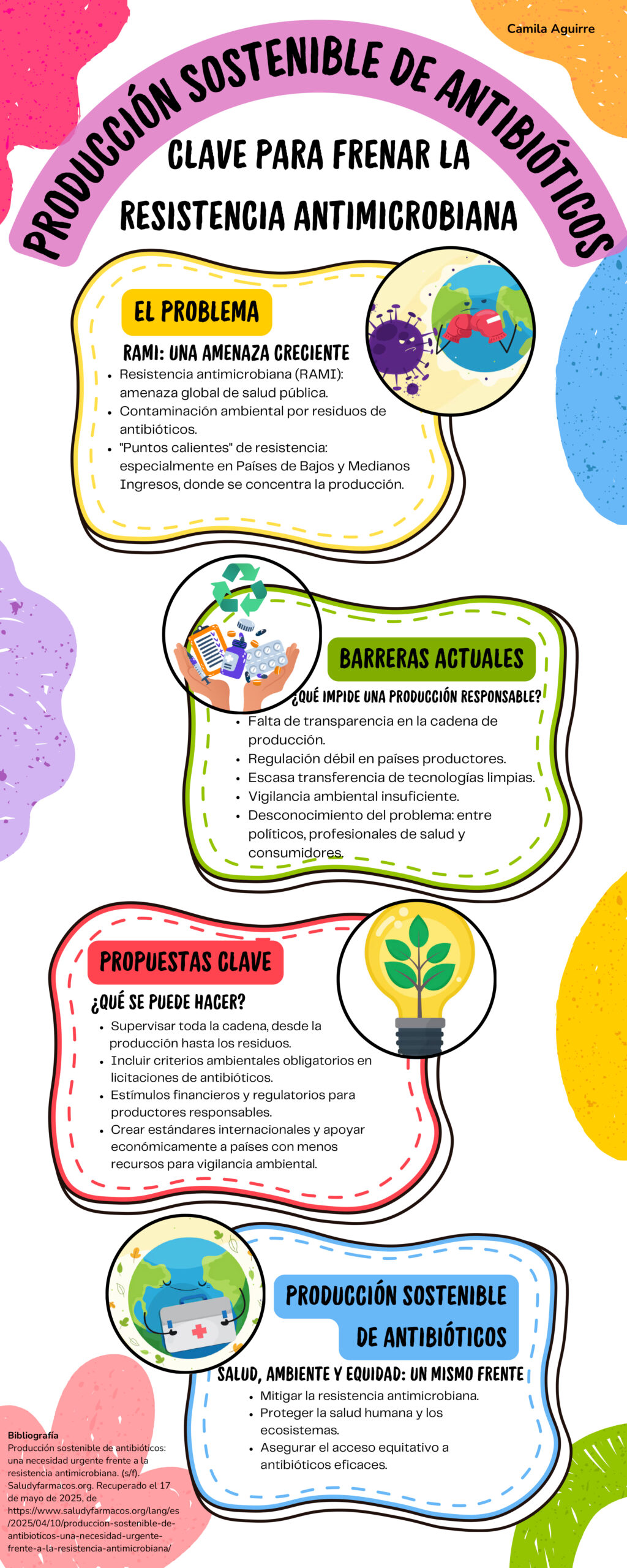
El 18 de junio de 2025, la FDA emitió una advertencia de seguridad para quienes usan el sistema transdérmico de escopolamina (transderm scop), un parche que se utiliza para tratar las náuseas y los vómitos, pues pueden experimentar complicaciones relacionadas con el calor [1]. El parche puede aumentar la temperatura corporal y disminuir la sudoración, lo que puede provocar complicaciones relacionadas con el calor, como confusión, pérdida del conocimiento, hospitalización o, en algunos casos, incluso la muerte.
La FDA recomienda que cualquier persona, especialmente los menores de 17 años y los adultos mayores de 60 años, que experimente síntomas de hipertermia como temperatura corporal elevada o disminución de la sudoración se retire el parche inmediatamente. Estas personas también deben evitar el uso de fuentes de calor externas como mantas térmicas, y deben mantenerse alejados de ambientes cálidos.
El sistema transdérmico de escopolamina, aprobado inicialmente en 1979, es un parche que se utiliza en adultos para tratar las náuseas y los vómitos asociados con el mareo por movimiento y la recuperación de la anestesia [2]. El parche para tratar las náuseas funciona liberando escopolamina, un anticolinérgico que bloquea las señales cerebrales responsables de las náuseas y los vómitos. El parche se aplica detrás de la oreja y libera un miligramo de escopolamina, cuyo efecto antiemético (evitar el vómito), comienza en cuatro horas y dura hasta tres días.
Las personas con glaucoma de ángulo cerrado o hipersensibilidad a la escopolamina no deben utilizar el parche. Aunque no está aprobado para su uso en niños, el sistema transdérmico de escopolamina se ha recetado ocasionalmente fuera de indicación para tratar la salivación excesiva en niños con parálisis cerebral u otros trastornos neurológicos [3].
La advertencia de seguridad sobre la escopolamina transdérmica se ha actualizado para añadir el riesgo de hipertermia a la información para la prescripción y al prospecto para el paciente. Los agentes anticolinérgicos como la escopolamina pueden elevar la temperatura corporal central y disminuir la sudoración. Estos efectos pueden agravarse por la exposición a fuentes externas de calor o en entornos con altas temperaturas. Estos síntomas, si no se detectan o no se tratan, pueden provocar complicaciones graves. En la mayoría de los casos la hipertermia se presenta dentro de las 72 horas posteriores a la aplicación. Incluso después de retirar el parche, los síntomas de abstinencia como mareos, dolor de cabeza y náuseas, pueden persistir durante varios días, ya que el medicamento absorbido permanece en el organismo [4].
La advertencia de la FDA se basa en 13 casos de hipertermia reportados a nivel global en pacientes que usan el parche para tratar las náuseas, incluyendo siete en EE UU. De estos casos, ocho involucraron a niños menores de 17 años y cuatro a adultos mayores de 60 años. Cuatro casos resultaron en hospitalización y dos pacientes murieron: uno era un niño y el otro un adulto mayor.
“En base al funcionamiento del medicamento, incluyendo su capacidad para atravesar la barrera hematoencefálica, una membrana que impide que sustancias potencialmente dañinas de la sangre lleguen al cerebro, la FDA determinó que existe evidencia razonable de asociación causal entre los parches de escopolamina y la hipertermia” [4].
El Grupo de Investigación en Salud de Public Citizen recomienda que, si utiliza el sistema transdérmico de escopolamina, esté atento a cualquier signo de calor excesivo o alteración de la temperatura, y que si presenta síntomas se retire el parche inmediatamente y se ponga en contacto con su médico.
Para consultar la alerta de la FDA, visite el siguiente enlace: https://www.fda.gov/media/187121/download
Informe cualquier evento adverso asociado con la escopolamina transdérmica (Transderm Scōp) al programa MedWatch de la FDA llamando al 1-888-463-6332 o visitando https://www.accessdata.fda.gov/scripts/medwatch/
Referencias