Nov
28
28/11/2025
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Crítica brecha entre el escrutinio de la FDA y la información que se publica sobre los ensayos clínicos
Salud y Fármacos
Boletín Fármacos: Ensayos Clínicos 2025; 28(4)
Tags: integridad de la ciencia, divulgación de datos primarios, olaparib, sotorasib y ciltacabtagene autoleucel, fallos metodológicos de los ensayos
Los debates recientes sobre la transparencia en los ensayos clínicos revelan una preocupación central: los médicos y la comunidad científica reciben información incompleta cuando dependen únicamente de los artículos publicados [1].
Expertos en regulación y ética sostienen que la FDA identifica riesgos, sesgos y fallos metodológicos que no figuran en muchos reportes académicos y publicaciones en revistas científicas sobre los mismos ensayos clínicos. Por otra parte, las publicaciones sobre los hallazgos de los ensayos clínicos, incluyendo los confirmatorios, influyen en las guías de práctica clínica, en el ejercicio médico y en las decisiones terapéuticas [2].
Si bien los procesos de revisión por pares suelen ser rigurosos, los revisores con frecuencia basan sus evaluaciones en datos secundarios divulgados por autores y patrocinadores, y este método es muy vulnerable vulnerabilidad, especialmente en el caso de los ensayos clínicos patrocinados por la industria farmacéutica, en cuyo caso los textos pueden no presentar una visión equilibrada, afirmaron investigadores del Centro de Ensayos Clínicos del Centro Nacional del Cáncer y del Hospital del Cáncer de la Academia China de Ciencias Médicas en Beijing [2].
El doctor Aaron Kesselheim, profesor en la Facultad de Medicina de Harvard y director del grupo del Programa de Regulación, Terapéutica y Derecho del Hospital Brigham and Women’s, propone que la FDA publique los resultados de sus revisiones de los datos primarios de los ensayos clínicos, y los análisis que presentan en las reuniones de los comités asesores y en las plataformas académicas para cerrar esa importante brecha de información [1]. A pesar de ser una propuesta de gran valor, el doctor Kesselheim duda que la agencia pueda asumirla por los drásticos recortes de personal, las restricciones presupuestales y la menor participación de los comités asesores externos que enfrenta actualmente el FDA.
Desde la Escuelo de Medicina de Yale, el doctor Joseph Ross respalda la importancia de difundir las revisiones independientes de la FDA porque a su juicio, esas evaluaciones contienen información que a veces contradice o matiza las narrativas de las empresas. Sin embargo, el doctor Ross advierte que las farmacéuticas no entregan sus bases de datos con la expectativa de que la agencia las publique en revistas científicas. Ross propone que la Biblioteca Nacional de Medicina indexe las revisiones regulatorias para que cualquier búsqueda sobre un medicamento redirija directamente a las evaluaciones de la FDA.
Investigadores del Centro Nacional del Cáncer de China fortalecen este argumento con ejemplos concretos. Compararon las publicaciones de los ensayos con las revisiones regulatorias de tres fármacos analizados por el Comité Asesor de Medicamentos Oncológicos (ODAC, por su sigla en inglés Oncologic Drugs Advisory Committee). Los casos evaluados fueron los fármacos olaparib, sotorasib y ciltacabtagene autoleucel. Los autores concluyeron que la FDA la FDA había detectado señales de riesgo, sesgos de diseño y problemas de ejecución que no aparecieron en los artículos revisados por pares.
Estas omisiones incluyeron heterogeneidad de la población del ensayo de olaparib donde se identificó también posibles peores resultados para pacientes con cáncer de próstata metastásico resistente a la castración sin variante BRCA.
En el caso de sotorasib los investigadores documentaron abandonos asimétricos, relecturas radiológicas que alteraron los resultados provisionales de supervivencia libre de progresión, un posible uso inadecuado del procedimiento de confirmación de progresión radiológica que alteró los resultados del estudio, cruces tempranos y evaluaciones de imágenes del investigador que favorecieron al grupo de sotorasib.
En cuanto a la reunión del ODAC sobre la terapia ciltacabtagene autoleucel, los investigadores destacaron varios problemas no identificados en los estudios publicados en revistas científicas, entre ellos, una mayor tasa de eventos adversos que condujeron a la muerte en los 90 días posteriores al inicio del tratamiento con ciltacabtagene autoleucel. así comouna tendencia a la disminución de la supervivencia global durante los primeros 10 meses posteriores al tratamiento.
Los autores señalan que estos patrones no representan incidentes aislados y resaltan que varios ensayos alcanzan los desenlaces principales, pero exhiben deficiencias que solo emergen cuando la FDA analiza los datos primarios. Los investigadores concluyen que la revisión por pares es necesaria, pero resulta insuficiente para evaluar la complejidad metodológica de los estudios pivotales.
Ante este panorama, los investigadores recomiendan que las revistas científicas exijan la publicación de los datos primarios de los ensayos confirmatorios, que los reguladores difundan activamente sus evaluaciones independientes y consideran que los médicos deben revisar de manera sistemática las opiniones regulatorias, ya que ofrecen un escrutinio más profundo que el proceso editorial tradicional.
En su análisis final, el doctor Kesselheim considera que “el siguiente paso lógico” es publicar las evaluaciones regulatorias independientes para fortalecer la integridad científica y la toma de decisiones clínicas.
Referencias:
Reconocimiento a la labor de 28 años construyendo comunidad por el acceso y uso adecuado de los medicamentos
Salud y Fármacos, 21 de noviembre de 2025
Tags: Acceso a medicamentos, uso adecuado de medicamentos, III Premios Asociación por el acceso justo a medicamentos, Salud y Fármacos, AAJM
El pasado 15 de noviembre el Ayuntamiento de Noblejas celebró la III edición de los Premios de la Asociación por un Acceso Justo al Medicamento (AAJM), donde se reconoció el trabajo de varias instituciones y profesionales comprometidos con el acceso a medicamentos.
En esta ocasión, la organización internacional Salud y Fármacos recibió el galardón a la Mejor Labor de una Organización, un reconocimiento que destaca sus 28 años de trayectoria en la generación y difusión de conocimiento sobre el acceso y el uso adecuado de medicamentos entre población hispanoparlante.
En 1997, un pequeño grupo de profesionales decidió unirse para enfrentar un problema que traspasaba fronteras: el acceso y el uso adecuado de los medicamentos. En 1998 Salud y Fármacos publicó el primer Boletín Fármacos y en el año 2005 se incorporó como organización sin ánimo de lucro en Texas, EE UU. Así nació Salud y Fármacos, un proyecto que empezó con dos boletines al año y que hoy produce ocho boletines trimestrales llenos de análisis independiente y de evidencia crítica bajo la lupa y el rigor metodológico de su fundador, Doctor Antonio Ugalde y su cofundadora y actual Directora Ejecutiva, Doctora Nuria Homedes.
Con el impulso y los recursos provistos por el Doctor Antonio Ugalde y el trabajo colectivo de universidades, organizaciones y activistas de América Latina, la red creció hasta convertirse en una verdadera biblioteca viva sobre las dinámicas e implicaciones de las políticas farmacéuticas a nivel global. Hoy, el buscador de la página web de https://www.saludyfarmacos.org/ permite acceder a un gran repositorio que alberga décadas de conocimiento y que es una herramienta clave para quienes trabajan por sistemas de salud más justos.
Salud y Fármacos es Miembro de la Sociedad Internacional de Boletines de Medicamentos (ISDB, International Society of Drug Bulletins) y obtuvo el nivel de reconocimiento Platinum Transparency 2025 que refleja su compromiso con la rendición de cuentas y con la transparencia.

En palabras de la Dra. Homedes durante el acto de premiación: ¨El principio de Salud y Fármacos sigue intacto: seguir diseminando información independiente, colaborativa y útil para los lectores, lejos de los intereses de la industria¨… ¨Este reconocimiento no solo celebra la labor de Salud y Fármacos, celebra a todas las personas, investigadores y organizaciones que siguen defendiendo el acceso equitativo al medicamento. Porque, frente a desafíos globales tan grandes, nadie sobra. Cuantos más seamos, mejor¨.

En el marco del evento de premiación, también se otorgó reconocimiento al Colegio Oficial de Médicos de Toledo, a la Confederación Salud Mental España, al especialista en farmacia hospitalaria Francesc Puigventós, a la periodista Sara Plaza Casares y al escritor y director de cine Roberto Santiago, quien recibió un reconocimiento especial por su libro: “La rebelión de los buenos” sobre las prácticas y abusos de la industria farmacéutica.
Comercialización ilegal de medicamentos en plataformas de comercio electrónico, sitios web y aplicaciones de dispositivos móviles
COFEPRIS, 31 de octubre de 2025
https://www.gob.mx/cms/uploads/attachment/file/1034684/Alerta_Sanitaria_Com._Ilegal_Med._31102025.pdf
La Comisión Federal para la Protección contra Riesgos Sanitarios (COFEPRIS) informa sobre la comercialización ilegal de medicamentos que se ofertan en plataformas de comercio electrónico, sitios web y aplicaciones de dispositivos móviles. Esta alerta se emite como resultado de las acciones de control sanitario realizadas por esta Comisión en las cuales se detectó la venta de medicamentos falsificados o sin registro sanitario, ofrecidos a precios inferiores a los del mercado y sin solicitar receta médica.
Para ver la lista de productos cuestionados puede ir al enlace que aparece en el encabezado.
Recomendaciones de COFEPRIS par la población:
La polifarmacia y el aumento de la mortalidad en adultos mayores
Salud y Fármacos
Tags: Caídas en el adulto mayor, polifarmacia y muertes en adultos mayores, lesiones por caídas en adultos mayores frágiles, riesgo de caídas en adultos mayores, opioides, benzodiacepinas, gabapentinoides, antidepresivos
Aunque el envejecimiento trae consigo problemas de equilibrio, dolores articulares, dificultades para la marcha, alteraciones visuales, auditivas y/o cognitivas que confieren especial fragilidad, estos factores aunados a la soledad y los entornos domésticos inseguros, no explican por sí solos el aumento de la mortalidad por caídas entre las personas mayores de 65 años que se ha documentado en EE UU durante las últimas décadas.
El problema no radica solo en la fragilidad física del envejecimiento o en los peligros cotidianos del hogar, todo apunta a un fenómeno más reciente. El detonante parece estar en el botiquín: la polifarmacia, especialmente con ciertos medicamentos, aumenta el riesgo de caídas en los adultos mayores.
Según un artículo publicado recientemente en JAMA Network, más de 41.000 adultos mayores fallecieron en EE UU durante el 2023, a causa de lesiones secundarias a caídas [1], una cifra que superó el número total de muertes por las siguientes causas: cáncer de mama o de próstata, las muertes derivadas de accidentes de tránsito, por sobredosis o cualquier otra lesión no intencional.
Lo más preocupante es que la tasa de mortalidad por caídas en este grupo etario se ha más que triplicado en las últimas tres décadas, mientras que en otros países de altos ingresos con poblaciones igualmente envejecidas han logrado reducirlas de manera sostenida.
En EE UU, el uso de medicamentos en adultos mayores está generalizado. Nueve de cada diez personas mayores de 65 años toman al menos un fármaco de venta con receta, casi la mitad (43%) consume varios fármacos simultáneamente, y el 45% utiliza medicamentos potencialmente inapropiados para su edad.
Muchos de los fármacos, conocidos como “medicamentos que incrementan el riesgo de caídas” (FRID, por su sigla en inglés Fall Risk–Increasing Drugs), pueden causar somnolencia, debilidad muscular, pérdida del equilibrio o dificultades para caminar, por lo que aumentan el riesgo de caídas. Entre ellos se encuentran los betabloqueadores, los anticolinérgicos, los inhibidores de la bomba de protones, los opioides, las benzodiacepinas, gabapentinoides y antidepresivos. Los cuatro últimos son los más peligrosos y todos actúan sobre el sistema nervioso central.
El incremento en las prescripciones de opioides comenzó en los años noventa, coincidiendo con el ascenso de las muertes por caídas. En la década siguiente, se disparó el uso de benzodiacepinas, y pronto se volvió habitual la combinación de opioides y sedantes, una mezcla particularmente peligrosa (Ver gráfico 1).
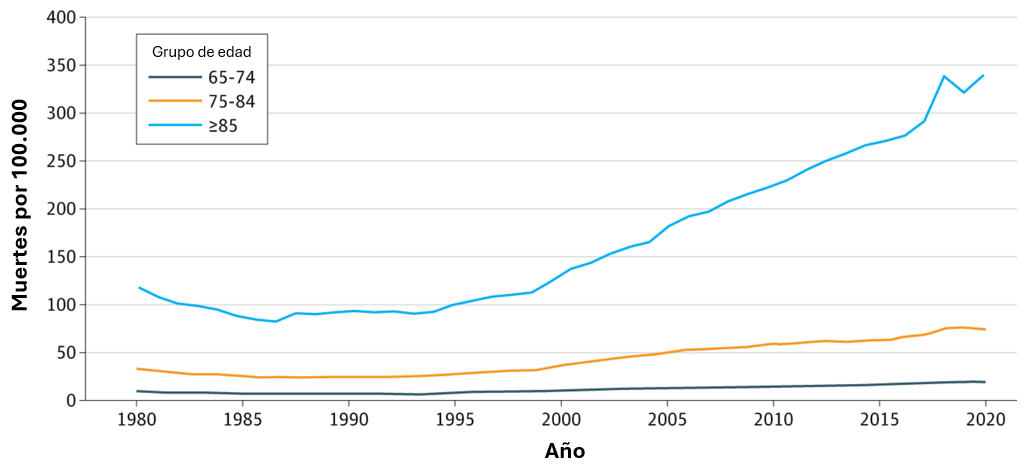
Aunque desde 2012 las prescripciones de opioides y benzodiacepinas han disminuido, su uso sigue siendo muy alto. Más recientemente, ha aumentado el uso de gabapentinoides como alternativa “más segura” para tratar el dolor crónico, pero su uso combinado con opioides se cuadriplicó entre 2006 y 2018. En paralelo, el consumo de antidepresivos en mayores de 65 años pasó del 8% al 20% en solo dos décadas.
Estas prácticas persisten a pesar de las advertencias de la Sociedad Americana de Geriatría, que desaconseja la prescripción de benzodiacepinas, antidepresivos y cualquier combinación de opioides con gabapentinoides o sedantes a los adultos mayores, y recomienda fuertemente evitar la prescripción de combinaciones de opioides con benzodiacepinas o gabapendinoides.
Resulta difícil justificar que casi una quinta parte de los adultos mayores de 85 años consuma benzodiacepinas, o que uno de cada cinco adultos mayores reciba antidepresivos, cuando la evidencia clínica indica que sus beneficios son limitados y sus riesgos sustanciales. Esta crisis debe ser reconocida y habría que tomar medidas contundentes y adaptadas a los diferentes contextos para disminuir la mortalidad prevenible y proteger la salud y la vida de los adultos mayores frágiles.
La solución no pasa solo por revisar las prescripciones de manera individual, se requiere una estrategia de salud pública que transforme la práctica médica, que aproveche los sistemas electrónicos de información para identificar a pacientes en riesgo, alertar a los prescriptores, ofrecer alternativas terapéuticas más seguras, involucrar a los farmacéuticos y establecer metas de calidad relacionadas con la seguridad farmacológica.
Si se lograra reducir la polifarmacia y el uso inadecuado de medicamentos en este grupo etario, en EE UU se podrían prevenir alrededor de 25.000 muertes anuales de adultos mayores por esta causa.
Fuente Original
La ausencia de incertidumbres y limitaciones de ensayos clínicos oncológicos en la evidencia publicada
Salud y Fármacos
Tags: limitaciones de ensayos clínicos oncológicos, incertidumbres no divulgadas en revistas científicas, limitaciones no reportadas en guías clínicas oncológicas
La aprobación acelerada de medicamentos oncológicos por parte de la FDA incrementó la probabilidad de introducir medicamentos con incertidumbres clínicas no resueltas, en base a los resultados de ensayos clínicos con limitaciones. La FDA aprueba más del 75% de los nuevos fármacos contra el cáncer a través de la vía acelerada. Es frecuente que la autorización de comercialización de los fármacos oncológicos se sustente en desenlaces no validados, estudios de un solo brazo y/o con insuficientes datos de seguimiento.
Aunque la agencia reguladora documenta detalladamente estas limitaciones en sus evaluaciones de beneficio-riesgo, dicha información rara vez llega a los clínicos, quienes habitualmente dependen de las publicaciones científicas y de las guías de práctica clínica para orientar sus decisiones terapéuticas.
Un estudio reciente identificó todos los medicamentos oncológicos aprobados por la FDA entre 2019 y 2022, y comparó las incertidumbres descritas por los evaluadores regulatorios con aquellas reportadas en los artículos de los ensayos pivotales y en las guías de la National Comprehensive Cancer Network (NCCN). Las aprobaciones de la FDA se otorgaron en base a los resultados de 56 ensayos pivotales.
Hasta abril de 2025, se habían publicado 51 ensayos para 48 fármacos contra el cáncer en revistas científicas y en las Guías referenciadas. De esos 48 fármacos oncológicos aprobados, según la evaluación beneficio-riesgo de la FDA, el 79% (n=38), presentaban incertidumbres clínicas relevantes (n=94); sin embargo, las publicaciones científicas solo mencionaron el 22% de estas incertidumbres y las guías clínicas NCCN solo el 23%.
Más de la mitad de los artículos sobre medicamentos con incertidumbres relevantes (20/38) y casi la mitad de las guías (18/38) omitieron todas las incertidumbres que había identificado la FDA.
Este hallazgo sugiere una brecha importante entre la evaluación regulatoria y la divulgación de información científica, con el riesgo de que los profesionales de la salud sobrestimen los beneficios y/o subestimen los riesgos de los nuevos fármacos aprobados contra el cáncer.
Los autores señalan que esta discrepancia puede estar relacionada con la ausencia de requerimientos específicos en las guías de publicación y en el desarrollo de guías clínicas.
A pesar de los avances recientes del CONSORT-Surrogate, que exige justificar el uso de criterios de valoración indirectos, persiste la falta de mecanismos sistemáticos para integrar las evaluaciones regulatorias en las recomendaciones clínicas.
Para fortalecer la transparencia y la toma de decisiones informadas, se propone que la FDA haga más accesibles sus informes sobre la evaluacion de beneficio-riesgo y que las revistas científicas, revisores y sociedades médicas adopten estándares que obliguen a declarar explícitamente las principales incertidumbres de los ensayos clínicos. Solo así podrá garantizarse que la práctica oncológica se base no solo en evidencia publicada, sino en una comprensión completa de sus limitaciones y sus numerosas incertidumbres.
Fuente original:
Cherla A, Wagner AK, Wouters OJ, et al. Reporting of Clinical Trial Uncertainties with new cancer drugs in Journal publications and Clinical Guidelines. JAMA. Published online September 03, 2025. doi:10.1001/jama.2025.13917.
Entrevista Ensayos clínicos en América Latina: Riesgos y dilemas éticos de la “Innovación científica”
Salud y Fármacos
En América Latina, la expansión de los ensayos clínicos patrocinados por la industria farmacéutica transnacional ha planteado serios interrogantes éticos y regulatorios.
Colombia se perfila como centro regional para esta investigación, en este panorama complejo y lleno de tensiones entre innovación, economía y bioética, proponemos una serie de reflexiones, que las presentamos a modo de entrevista. Natalia Castrillón entrevista a Nuria Homedes (NH) y a Bernardo Useche (BU).
NH. Se desconoce el número de ensayos clínicos que a nivel global se inician en un año determinado, pero son miles. Solo en Colombia cada año se registran más de 60 ensayos clínicos financiados por la industria farmacéutica. También sabemos que las agencias reguladoras de referencia (FDA, EMA), aprueban – en un buen año- 40 o 50 productos nuevos, pero de esos – según las agencias reguladoras y los boletines independientes de medicamentos- solo uno o dos añaden valor terapéutico a los tratamientos existentes.
Hay años en que ninguno de los medicamentos recién aprobados es superior al tratamiento estándar, y hay años en que se produce un medicamento verdaderamente revolucionario. Lo que sí suele suceder es que estos medicamentos nuevos son más caros que los existentes.
Otra cosa a tener en cuenta es que, según datos de la industria, solo alrededor del 10% de los productos que se testan en ensayos clínicos en humanos (Fase I a Fase III) acaban siendo aprobados por las agencias reguladoras de referencia. Es decir, muchas moléculas se quedan por el camino. Estos porcentajes varían por especialidad (son más positivos para enfermedades respiratorias, pero peores en el caso de productos oncológicos). Estas cifras podrían mejorar con el uso de farmacogenómica.
Ahora bien, la mayoría de los ensayos clínicos que se hacen en AL son de Fase III, y el porcentaje de éxito de estos ensayos es de alrededor del 50%, por lo que podemos asumir que solo la mitad de los productos experimentales que se testan en ensayos de Fase III en América Latina serán aprobados por las agencias reguladoras de referencia – y si se tiene en cuenta que solo la mitad de los participantes en los ensayos clínicos reciben el medicamento experimental (la otra mitad suele recibir el tratamiento habitual o un placebo) pues queda claro que solo una parte de los participantes en ensayos clínicos se benefician.
También hay que tener en cuenta que algunos efectos adversos de los medicamentos no se conocen hasta que aumenta el número de personas que los utilizan, por eso hay quienes recomiendan que no se consuman medicamentos nuevos hasta 7 años después de su aprobación, para dar tiempo a que se conozca su perfil de seguridad [1]. La información disponible indica que la mayoría de los productos que resultan ser inseguros cuando los utiliza la población general, se retiran del mercado durante ese periodo de tiempo.
NH, El riesgo más importante para los participantes es que se está utilizando un producto experimental. Es decir, los participantes son parte de un experimento. Esta palabra ha desaparecido del léxico que se utiliza al hablar de ensayos clínicos, y no solo en los países hispanoparlantes [2], por las connotaciones negativas que puede tener el decir que se está experimentando en humanos. Solemos referirnos a los ensayos clínicos como protocolos, proyectos, estudios…
El otro riesgo que identificó el personal de la agencia reguladora de ensayos clínicos de Perú es que los participantes en los ensayos clínicos no siempre se adhieren a las indicaciones del investigador principal o del coordinador del estudio. No lo hacen con mala intención, al contrario, lo hacen para no molestar y por ignorancia. Desconocen la importancia de adherirse a las instrucciones y cómo esto afecta los resultados… y a veces, si se sienten mal después de tomar el medicamento por la noche o los fines de semana, para no molestar al equipo de investigación no dicen nada [3].
Lo que la agencia reguladora de Perú descubrió es que muchos de los participantes en los ensayos clínicos consumían otros medicamentos o productos de medicina tradicional y no se lo decían al médico investigador. A veces se iban a las salas de emergencia de hospitales y no les decían que estaban participando en un ensayo clínico, otras veces el participante tomaba la mitad de la dosis… Esta información no siempre la compartían con el investigador, porque no querían que los echaran del “proyecto” (protocolo) [4].
Hay que decir que los participantes en ensayos clínicos suelen estar muy satisfechos con el trato que reciben, con la privacidad con la que se les trata, y con las pruebas que les hacen… Se sienten bien cuidados… y no son conscientes de los riesgos que conlleva el producto experimental o las pruebas clínicas que se les realizan.
NH. Si, el consentimiento informado lo dice, pero muchos participantes en ensayos clínicos firman el consentimiento informado sin haberlo leído, y si lo han leído es reconocido que no lo entienden, hacen lo que se les dice porque se fían de su médico…y otras veces hay una inducción indebida por parte del reclutador. En el estudio de Perú algunos entrevistados dijeron: ¨el médico me dijo que si fuera su hija me inscribiría en el ensayo… que si participaba en el ensayo me curaría, … que si participaba en el ensayo tendría acceso al mejor tratamiento, si no, solo al del sector público¨ [5].
Los participantes a veces leen el consentimiento cuando llegan a su casa, pero los consentimientos son cada vez más largos, incluyen términos que los participantes no entienden, incluso miembros de CEI dicen que no los entienden. Es muy difícil transmitir a los participantes todo lo que deben de saber en un solo documento.
La mayoría de los entrevistados en Perú sabían que si se sentían mal tenían que ir al centro de investigación, pero no lo hicieron por no molestar o porque no asociaron su problema con la participación en el ensayo.
NH. La regulación de la mayoría de los países de América Latina exige que los CEI den seguimiento a los participantes en ensayos clínicos, pero eso se hace en base a los documentos que les entrega el investigador principal, el patrocinador o las agencias reguladoras. No hablan con los participantes, por lo que no tienen acceso a información importante como la que descubrió la agencia reguladora de Perú.
Solo dos hospitales de una provincia de Buenos Aires han contratado a trabajadoras que verifican que los participantes entiendan el consentimiento informado antes de firmarlo, pero después no dan seguimiento al paciente. Este seguimiento del paciente es muy importante para asegurar la integridad de la información que se recaba durante los ensayos clínicos, y también para proteger a los participantes. Hay CEI que tienen interés en este tipo de actividad, pero no tienen tiempo para hacerlo. Además es una actividad que no está remunerada.
NH. Esto depende de lo que diga el protocolo. Hay muchas metodologías para tratar de documentar la adherencia al tratamiento. El asunto es que el participante quiere complacer al investigador porque en general los participantes quieren permanecer en el estudio… eso hace que el paciente no siempre comparta toda la información con el investigador… Los que fueron entrevistados en el estudio de Perú contaron cosas, pero dijeron a los entrevistadores, “por favor no se lo diga al investigador porque me echará del estudio”.
La persona que entreviste a los participantes tiene que hacerse amiga de ellos, tiene que ser alguien en quien ellos sientan que pueden confiar, que no los va a delatar y que se preocupa por su seguridad y por su salud.
NH. Hace unos años, 2015 y 2016 revisamos los medicamentos que la FDA había aprobado en 2012 que se habían testado en América Latina, eran 33, y nos encontramos con que un 30% no se habían registrado ni comercializado en ninguno de los países latinoamericanos donde se habían testado… Simplemente las empresas farmacéuticas no tenían interés en comercializar esos medicamentos en esos países y no los registraron. A veces los registraron, pero no los comercializaron porque esperaban a que las condiciones del mercado fueran más favorables [6].
Solo el 25% de los medicamentos (n=8) se comercializaron en todos los países en que se habían testado. Entre los medicamentos que se comercializaron, solo uno costaba menos de un salario mínimo mensual, la inmensa mayoría costaban más de cinco salarios mínimos mensuales – y en un caso hasta 899 veces.
Además, los boletines independientes de medicamentos habían estudiado la ventaja comparativa de 26 de estos medicamentos nuevos con los tratamientos existentes, 10 de ellos habían sido clasificados como No Usar, solo cinco de ellos se clasificaron como posiblemente superiores para poblaciones especiales, generalmente reducidas, y solo tres de ellos se habían comercializado en los países donde se habían testado. Es decir, la mayoría de los medicamentos que se testaron en América Latina, cuando se comercializaron, resultaron ser inasequibles para la mayoría de la población y de los sistemas de salud latinoamericanos, además la mayoría no son superiores a los tratamientos existentes [7].
NH. Las debilidades que identificamos en América Latina no son específicas de la región… También están presentes en países de altos ingresos. El primer problema es que se puede constituir un CEI con solo 5 miembros, en la mayoría de los casos los CEI son más grandes, 7-15 personas, para garantizar quorum. Esos CEI revisan todo tipo de proyectos, desde ensayos clínicos con medicamentos financiados por la industria a tesis de estudiantes y ensayos con dispositivos médicos.
Además, suele ser una tarea mal remunerada y con frecuencia los miembros trabajan ad honorem. Como resultado, no todos los CEI que revisan protocolos de ensayos clínicos con medicamentos cuentan con miembros expertos en el área clínica en la que se va a aplicar el producto en investigación, ni con metodólogos especializados en ensayos clínicos. Es decir, si bien esos CEI pueden evaluar tesis de estudiantes, muchos de ellos no cuentan con la capacidad técnica necesaria para evaluar los diseños de los ensayos clínicos que les presenta la industria y que suelen estar muy bien escritos.
Los CEI tienen la posibilidad de consultar con expertos, pero pocos lo hacen.
Otro problema es que los ensayos clínicos son un negocio…. Eso es algo que no aparece en las formas de consentimiento informado pero que se discute si se debería incluir. Tanto los investigadores principales como los centros donde se realiza la investigación se benefician económicamente de los ensayos clínicos que realizan. Muchos de los CEI incluyen a miembros que hacen o han hecho ensayos clínicos financiados por la industria, y tienen un sesgo – no siempre reconocido- por favorecer a sus colegas… “Hoy por ti, mañana por mi” … También se sabe que a veces los administradores de los hospitales o centros de investigación imponen sus deseos de que se realicen ensayos clínicos.
Hay que resaltar que la mayoría de CEI no tienen acceso a los contratos entre el patrocinador y el investigador y/o el centro de investigación, por lo que no pueden evaluar si hay alguna cláusula del contrato que puede inducir a que se violen los criterios de inclusión en un ensayo clínico o a que se retenga a un paciente que debería retirarse del ensayo.
El resultado final es que los CEI suelen aprobar los ensayos clínicos que presenta la industria, lo que contrasta con las afirmaciones que han hecho metodólogos de renombre sobre la calidad del diseño de muchos de dichos ensayos clínicos.
Los países compiten en aprobar lo más rápido posible los ensayos de la industria, para conseguir más ensayos. Los miembros de los CEI que entrevistamos en diferentes países de América Latina (incluyendo Argentina, México, Costa Rica, Panamás, Perú y Colombia) dijeron que cuando evalúan ensayos clínicos financiados por la industria no solicitan cambios, porque si lo hacen se retrasaría el reclutamiento de pacientes y esto perjudicaría al investigador y a la institución [8].
En resumen, la mayoría de los CEI, no todos, suelen aprobar los ensayos clínicos financiados por la industria, la proporción de estudios que rechazan es muy baja y suele deberse a problemas administrativos.
Se puede pensar que los países/regiones tendrían que profesionalizar a los CEI que reciben los ensayos clínicos patrocinados, para que los evalúen expertos en la materia. Eso no eliminaría a los CEI institucionales, quienes podrían aceptar o no aceptar el dictamen del CEI especializado, y quienes se responsabilizarían de monitorear y proteger a los participantes.
NH. Bueno, es que pienso que no se debería consolidar. No le veo las ventajas de hacerlo…
Agencias internacionales y países están afirmando que es importante que se hagan ensayos clínicos para: (1) dinamizar la economía; (2) desarrollar la capacidad de investigación. Sin embargo, no hemos encontrado evidencia para sustentar dichos argumentos.
Según la misma industria, en el 2016, por cada ensayo clínico que se hace en un país latinoamericano ingresan entre uno y dos millones de dólares… En el caso de Colombia esto representaría alrededor de US$140 millones (quizás el doble si se tienen en cuenta las externalidades) [9], pero eso no son ingresos netos, pues hay que descontar los gastos en los que incurre el INVIMA en la aprobación y supervisión de los ensayos clínicos, así como los gastos de los CEI.
También habría que tener en cuenta los gastos médicos de las personas que participan en ensayos clínicos y experimentan eventos adversos, que con frecuencia corren a cargo del erario público o de las aseguradoras, así como parte del tiempo del personal de salud que reclutan a los pacientes para los ensayos y tienen que verificar si cumplen los criterios de inclusión. Es decir, hay gastos asociados a la realización de los ensayos clínicos que en muchos casos no los pagan los patrocinadores, sino que corren a cargo del sector público o de las aseguradoras, o los absorbe el personal que dona su tiempo.
Que nosotros sepamos, nadie ha contabilizado lo que los países invierten para que la industria realice los ensayos clínicos, por lo que se desconoce cuál es el aporte neto de la industria de los ensayos clínicos a la economía colombiana o a la de cualquier otro país.
El otro argumento es que los ensayos clínicos contribuyen al desarrollo científico. Tenemos muchos problemas con esta afirmación, porque lo que los científicos deben aprender es a diseñar proyectos de investigación y analizar la información recabada… La gran mayoría de los que hacen ensayos clínicos patrocinados por la industria no participan de las discusiones sobre el diseño de la investigación ni del análisis de los datos, simplemente recaban datos… Eso no es capacitar en investigación nada más administran un proyecto diseñado en un país de altos ingresos, en donde también se analizarán los datos recabados en Colombia. En Colombia solo se recaban datos. Para mejorar las capacidades de investigación habría que invertir en becas de estudios doctorales, ya sea en Colombia o en el extranjero.
BU. La Doctora Homedes hizo un análisis en el que se revela información importante y poco conocida sobre los ensayos clínicos de las multinacionales farmacéuticas. Especialmente dos puntos llaman la atención:
El caso en Colombia de Carolina Jiménez en el cual, sin atender lo firmado en el consentimiento informado, no se informó a la participante de los resultados del ensayo clínico que comprometían en materia grave su salud, es ilustrativo [10].
En Colombia, cada vez son más los ensayos clínicos de la industria en busca de nuevos medicamentos que puedan patentar, dadas las ventajas que les otorgan los Tratados de libre Comercio con Europa y EE UU. Más aun, el gobierno de Gustavo Petro mediante reciente decreto presidencial mantendrá las normas de propiedad intelectual vigentes en los tratados de libre comercio [11].
Según el informe de la Asociación Colombiana de Centros de Investigación Clínica (ACIC) del 2024, 160 centros de investigación tenían en curso 332 ensayos clínicos a los que se adicionaron ese año otros 29 estudios por un valor de US$73 millones. De esos 361 experimentos, 89 ensayos clínicos fueron patrocinados por Merk Sharp & Dohme, 37 por Novartis, 27 por AstraZeneca y 19 por Bristol Myers [12].
Según la Asociación de Laboratorios Farmacéuticos de Investigación y Desarrollo (AFIDRO), a 28 de agosto 2025, ya son mil los estudios clínicos en Colombia [13].
Las multinacionales farmacéuticas, a través de AFIDRO, quieren avanzar y “consolidar a Colombia como un hub regional de investigación” en América Latina. En mi opinión, con esta estrategia, la industria tiene como objetivos ampliar su posición predominante en un mercado creciente de los adultos mayores, competir con China, país que se ha posicionado como potencia mundial en investigación clínica [14] y compensar la pérdida de rentabilidad que supone el control de precios impuesto en EE UU por el presidente Trump a través de varias de sus órdenes [15] presidenciales [16] ejecutivas [17].
Parte integral de esta estrategia de la industria es hacer lobby para que los gobiernos latinoamericanos aumenten el presupuesto de salud. Según la directora ejecutiva de la Asociación Colombiana de la Industria Farmacéutica (ASCIF), en Colombia, los medicamentos ya representan entre el 25 y el 30% de la Unidad de Pago por Capitación (Nota de SyF: UPC es el valor que el Estado colombiano paga por cada ciudadano, este valor es diferencial según ciclo de vida, según régimen de afiliación al sistema de seguridad social -régimen contributivo y subsidiado- y según área geográfica).
Si se tiene en cuenta que los productores de medicamentos patentados tienen como meta acceder a un mayor porcentaje de los recursos públicos, se puede prever que en la medida en que se haga realidad esta proyección, la sostenibilidad financiera del sistema de salud colombiano se verá seriamente afectada.
No hay duda de que las multinacionales deben continuar siendo un jugador clave en el mercado farmacéutico en Latinoamérica, pero se convierte en una necesidad fundamental fortalecer la industria nacional. Es un problema de seguridad y soberanía farmacéutica.
[1] Sidney M Wolfe. The seven-year rule for safer prescribing. Aust Prescr 2012;35:138-9 https://australianprescriber.tg.org.au/articles/the-seven-year-rule-for-safer-prescribing-1.html
[2] Elliott, C. Whatever Happened to Human Experimentation? Hastings Center Report, 2016;46: 8-11. https://doi.org/10.1002/hast.531
[3] Minaya G, Fuentes D, Ugalde A, Homedes N. A missing piece in clinical trial inspections in Latin America: interviews with research subjects in Peru. Journal of Empirical Research on Human Research Ethics; 2017;12(4) 232–245. https://pubmed.ncbi.nlm.nih.gov/28728496/
[4] ibid
[5] Ibid
[6] Homedes N, Ugalde A. Ensayos clínicos en América latina: implicancias para la sustentabilidad y seguridad de los mercados farmacéuticos y el bienestar de los sujetos. Salud Colectiva 2016; 12(3): 317-345 https://revistas.unla.edu.ar/saludcolectiva/article/view/1073
[7] Ibid
[8] Puede leer los resultados de estas entrevistas en https://www.saludyfarmacos.org/publicaciones/informes/
[9] Pugatch Consilium. Challenges and Opportunities -Developing the biotechnology sector in Colombia. 2016. https://www.pugatch-consilium.com/reports/Challenges%20and%20Opportunities_v6.pdf
[10] Palacios C. La Country me usó como conejillo. El Tiempo, 29 de agosto de 2025. https://www.eltiempo.com/opinion/columnistas/la-country-me-uso-como-conejillo-3485283
[11] Ministerio de Salud y Protección Social. Decreto Presidencial 858 de 2025, artículo 2.11.2.4.2. Mecanismos de participación para la investigación, desarrollo y producción nacional de Tecnologías Estratégicas en Salud. https://www.funcionpublica.gov.co/eva/gestornormativo/norma.php?i=261736
[12] Asociación Colombiana de Centros de Investigación Clínica (ACIC). Resumen de la Medición del aporte económico de la investigación clínica en Colombia, Estudio 2024. https://aciccolombia.org/wp-content/uploads/2025/03/INFOGRAFIA.pdf
[13] Asociación de Laboratorios Farmacéuticos de Investigación y Desarrollo (AFIDRO). La investigación clínica. Consultado 28-08-2025. https://afidro.org/la-investigacion-clinica/
[14] Bruckner T. New study shows that China is now a global powerhouse of clinical research. TranspariMed, 26 de mayo de 2025. https://www.transparimed.org/single-post/new-study-shows-that-china-is-now-a-global-powerhouse-of-clinical-research
[15] Presidency Executive Order 14273 of April 15, 2025; https://www.federalregister.gov/documents/2025/04/18/2025-06837/lowering-drug-prices-by-once-again-putting-americans-first
[16] Presidency Executive Order 14293 of May 5, 2025; https://www.federalregister.gov/documents/2025/05/08/2025-08267/regulatory-relief-to-promote-domestic-production-of-critical-medicines
[17] Presidency Executive Order 14297 of May 12, 2025; https://www.federalregister.gov/presidential-documents/executive-orders/donald-trump/2025
Valoraciones que hizo Prescrire de medicamentos nuevos durante 2024: una breve revisión
(Prescrire’s ratings of new drugs in 2024: a brief review)
Prescrire International 2025; 34 (269): 102-104
Traducido por Salud y Fármacos, publicado en Boletín Fármacos: Prescripción, Farmacia y Utilización 2025; 28 (3)
Tags: avance terapéutico de nuevos fármacos, indicaciones nuevas con pocos beneficios, nuevos permisos de comercialización más peligrosos que beneficiosos, nuevos fármacos no ofrecen ventajas sobre otros tratamientos existentes
Todos los meses, Prescrire publica revisiones sistemáticas independientes de adelantos recientes en el mercado farmacéutico europeo, incluyendo los permisos de comercialización otorgados a nuevos principios activos, nuevas indicaciones, nuevas formas farmacéuticas, etc.
Nuestro objetivo es ayudar a nuestros suscriptores a identificar aquellos que mejoren el cuidado del paciente. También estamos atentos a las reevaluaciones del balance riesgo-beneficio de medicamentos que ya están en el mercado, a las novedades en cuanto a sus efectos adversos, a la escasez de medicamentos y a los retiros del mercado.
En general, menos avances terapéuticos reales que en años anteriores.
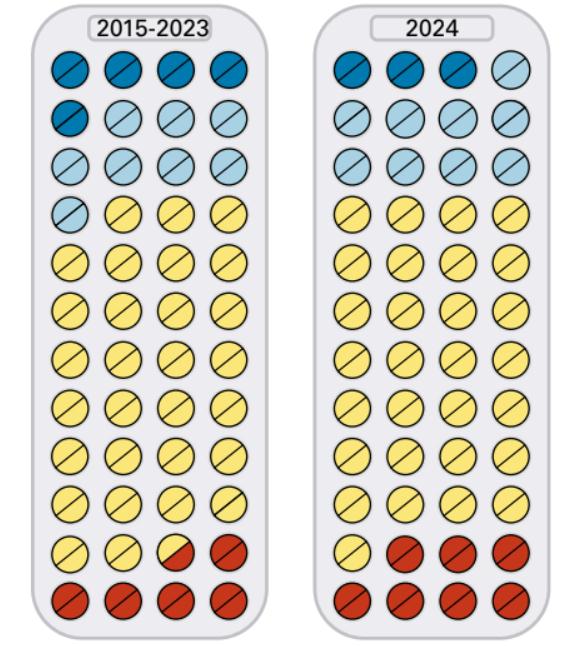
Durante 2024 Prescrire evaluó 122 permisos de comercialización nuevos, solo 30 de estos añadieron algún beneficio a los que aportan los tratamientos existentes. De los 30, uno (el 0,8% de todos los permisos de comercialización nuevos que evaluamos en nuestra edición en francés durante 2024), representó un avance terapéutico importante (que valoramos como “bravo”): el fexinidazol (Fexinidazol Winthrop) para la enfermedad del sueño causada por el Trypanosoma brucei rhodesiense. Otros seis (el 5% de nuestras evaluaciones), representaron un avance notable (que valoramos como “ofrece una ventaja”) y los otros 23 (el 19%), representaron un avance mínimo (que valoramos como “puede ser de ayuda”).
El número de indicaciones o productos nuevos que representaron al menos un avance notable (que evaluamos como “ofrece una ventaja”) estuvo por debajo del promedio que observamos durante los nueve años anteriores.
Casi la mitad de los nuevos permisos de comercialización que analizamos en 2024 (60, o el 49%) no ofrecía ventajas sobre otros tratamientos existentes (los valoramos como “nada nuevo”).
En 14 casos (el 11%), los datos no eran concluyentes (los valoramos como “se reserva la valoración”) y 18 permisos de comercialización nuevos (el 15%) se consideraron más peligrosos que beneficiosos para el problema clínico en cuestión (los valoramos como “no es aceptable”), una proporción más alta que la observada en los últimos años.
Algunos avances prácticos para los niños.
Dos novedades representaron un claro avance práctico para los niños:
Otros avances ofrecieron una pequeña ventaja práctica, en particular los comprimidos bucodispersables con 5mg de dolutegravir + 60 mg de abacavir + 30 mg de lamivudina (Triumeq) para los niños infectados por VIH que pesan entre 14 y 25kg, lo que elimina la necesidad de usar dos o hasta tres productos diferentes. También, las cápsulas duras con 100 mg de trientina (Cufence), que son útiles para ajustar las dosis diarias de algunos pacientes con enfermedad de Wilson.
Inhibidores de la Janus quinasa: por lo general, es mejor evitarlos para tratar las enfermedades inflamatorias crónicas.
La eficacia de los cuatro inhibidores de la Janus quinasa para tratar las enfermedades inflamatorias crónicas que evaluamos este año fue muy deficiente o no estuvo bien demostrada para justificar la exposición de los pacientes a sus efectos adversos cardiovasculares (incluyendo trombosis venosa y arterial) y a un aumento del riesgo de infecciones graves y de cáncer.
Los medicamentos y las enfermedades en cuestión son los siguientes:
el baricitinib y el ritlecitinib para la alopecia areata;
el upadacitinib para la enfermedad de Crohn y para la espondiloartritis axial “no radiográfica” y
el tofacitinib para la espondilitis anquilosante (espondiloartritis axial “radiográfica”).
Permisos de comercialización condicionales: las empresas presionan a la EMA.
En 2024 la EMA publicó conclusiones justificadamente desfavorables sobre la renovación de tres permisos de comercialización condicionales. Este tipo de permiso de comercialización se otorga en base a datos incompletos para cubrir una “necesidad médica insatisfecha” mientras se espera obtener datos adicionales, a menudo sobre la eficacia del medicamento.
En los tres casos, la presunta eficacia del medicamento no se confirmó con los datos obtenidos desde su entrada al mercado. Los medicamentos fueron los siguientes:
el atalureno (Translarna) para la distrofia muscular de Duchenne;
el belantamab mafodotina (Blenrep) para el mieloma múltiple, que se retiró del mercado a comienzos de 2024 y el ácido obeticólico (Ocaliva) para la colangitis biliar primaria [1-3].
En el caso del atalureno, la Comisión Europea no revocó el permiso de comercialización del medicamento y tomó la peculiar decisión de solicitar que la EMA reanalizara los datos. La EMA reafirmó su decisión tanto a mediados de 2024 como en octubre de 2024, tras otra solicitud de la farmacéutica en cuestión. El 3 de marzo de 2025 la Comisión Europea aún no había publicado su decisión definitiva [2].
En cuanto al ácido obeticólico, la Comisión Europea revocó su permiso condicional de comercialización, pero el Tribunal de Justicia Europeo suspendió temporalmente la decisión en septiembre de 2024: la farmacéutica sostuvo que la EMA no había tomado en cuenta “su abundante evidencia positiva según los datos de la práctica clínica (real world)” [3].
Finalmente, la revocación de este permiso de comercialización se confirmó en diciembre de 2024 [4]. El ácido obeticólico ha estado en la lista de medicamentos a evitar de Prescrire desde el 2019 ya que suele exacerbar los síntomas principales de la enfermedad (prurito y fatiga), y es posible que provoque efectos adversos hepáticos graves que en ocasiones son mortales [5].
Un cuarto caso es el permiso condicional de comercialización del pralsetinib (que antes se comercializaba bajo la marca comercial Gavreto), que se revocó a finales de 2024 a pedido de la farmacéutica, tras observarse un aumento de la incidencia de infecciones graves en un ensayo clínico (que se publicará en un número próximo).
El permiso de comercialización de un quinto medicamento, el darvadstrocel (que antes se comercializaba como Alofisel), que autorizaron precipitadamente para tratar las fístulas perianales, se revocó a finales de 2024 a pedido de la farmacéutica, debido a los resultados de un segundo ensayo clínico [6].
|
VALORACIÓN DE PRESCRIRE |
2015 |
2016 |
2017 |
2018 |
2019 |
2020 |
2021 |
2022 |
2023 |
2024 |
TOTAL |
|
BRAVO |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
1 |
2 (0,19) |
|
UN AVANCE REAL |
3 |
1 |
1 |
2 |
1 |
2 |
3 |
0 |
0 |
0 |
13 (1,22) |
|
OFRECE UNA VENTAJA |
5 |
5 |
9 |
11 |
10 |
6 |
14 |
11 |
10 |
6 |
87 (8,19) |
|
PUEDE SER DE AYUDA |
15 |
9 |
18 |
22 |
13 |
18 |
19 |
23 |
20 |
23 |
180 (16,95) |
|
NADA NUEVO |
43 |
56 |
45 |
50 |
61 |
55 |
51 |
63 |
73 |
60 |
557 (52,45) |
|
SE RESERVA LA VALORACIÓN |
6 |
5 |
4 |
5 |
9 |
17 |
12 |
13 |
10 |
14 |
95 (8,95) |
|
NO ES ACEPTABLE |
15 |
16 |
15 |
9 |
14 |
10 |
9 |
14 |
8 |
18 |
128 (12,05) |
|
TOTAL |
87 |
92 |
92 |
99 |
108 |
109 |
108 |
124 |
121 |
122 |
1.062 (100) |
BRAVO
OFRECE UNA VENTAJA
PUEDE SER DE AYUDA
NO ES ACEPTABLE
Proporción de autorizaciones en 2024 que mejoraron el cuidado del paciente, en comparación con los nueve años anteriores
 Avance notable
Avance notable Avance mínimo
Avance mínimo Sin ventajas probadas
Sin ventajas probadas Más peligroso que beneficioso
Más peligroso que beneficiosoMedicamentos que son más peligrosos que beneficiosos: uno menos en el mercado y ¿se retirará otro?
El permiso de comercialización europeo del ulipristal de 5 mg (Esmya), que se usa para tratar algunos casos de miomas uterinos, se revocó finalmente a mediados de 2024, a pedido de la farmacéutica. Los datos de la eficacia del uso continuo del ulipristal para este problema clínico no fueron concluyentes. Sin embargo, este producto expone a las pacientes a un riesgo de lesión hepática mortal.
El 30 de diciembre de 2024, aún se estaba realizando una revisión del balance riesgo-beneficio de Mysimba [la combinación de bupropión + naltrexona], solicitada por la Comisión Europea. La farmacéutica no cumplió su obligación de ejecutar un ensayo clínico post autorización para evaluar los efectos adversos cardiovasculares a largo plazo de esta combinación, autorizada para la pérdida de peso.
Este producto tiene efectos adversos cardiovasculares y neuropsiquiátricos desproporcionados, dada su eficacia limitada. Ya era hora de que se lo retirara del mercado. Los dos fármacos de esta combinación han estado en la lista de medicamentos a evitar de Prescrire durante años [5].
En resumen
Además de un avance terapéutico importante y algunos avances prácticos para niños, el 2024 no fue un buen año. Aproximadamente un séptimo de los permisos de comercialización nuevos que evaluamos eran opciones peores para los pacientes que las que ya existían, lo que es alarmante, sobre todo debido a que las revocaciones y las retiradas son escasas y casi siempre lentas.
Comentario de Salud y Fármacos
Tras una década de evaluaciones rigurosas, sistemáticas y metodológicamente sólidas sobre los permisos de comercialización de nuevos fármacos, Prescrire International publicó los resultados de la valoración acumulada cuyos resultados constituyen una seria alerta para las políticas farmacéuticas, la elaboración de formularios, la farmacovigilancia y la salud pública.
Los datos que resume el Cuadro 1 muestran que más del 52% (n=557) de los medicamentos aprobados en la última década no aportan beneficios clínicos adicionales a las terapias existentes para las mismas indicaciones, lo que cuestiona la pertinencia de su incorporación en los sistemas sanitarios, especialmente en entornos de recursos limitados, porque agrava la presión financiera.
Más grave aún es que la evidencia acumulada muestra que el 12% (n=128) de los nuevos fármacos aprobados que fueron evaluados, conllevan más riesgos que beneficios para los pacientes, configurándose como una falla crítica y reiterada de los mecanismos que deberían velar por proteger los derechos fundamentales a la salud y la vida; y representa una asignación de recursos mal direccionada e ineficiente.
La aprobación y comercialización de medicamentos con escaso o nulo valor terapéutico o peor aún, con perfiles de riesgo desfavorables, compromete la seguridad de la población y la expone a la incertidumbre de eventos adversos graves (que incluyen la muerte) y desvía recursos que podrían destinarse a intervenciones seguras y costo-efectivas que respondan a los problemas prioritarios de las comunidades.
Esta contundente evidencia acumulada por Prescrire (Ver cuadro 1), tras una década de aplicar rigor científico y metodológico a la evaluación de los nuevos fármacos aprobados por las agencias reguladoras , demuestra también que menos del 10% de los medicamentos evaluados ofrecen una ventaja o un avance real y menos del 17% de los medicamentos evaluados podrían ser de ayuda, lo que debería movilizar las voluntades políticas y a los interesados en políticas farmacéuticas, a exigir medidas más estrictas y libres de conflictos de intereses en los procesos de aprobación y regulación de nuevas tecnologías en salud (que en gran parte se pagan con recursos públicos).
Actuar con determinación y contundencia implica fortalecer la independencia de las agencias regulatorias, exigir comparaciones clínicas directas con tratamientos estándar, priorizar el beneficio a los pacientes y la transparencia en los procesos de aprobación, y fortalecer los sistemas de farmacovigilancia post comercialización. Un buen sistema de farmacovigilancia permitiría detectar y retirar de manera oportuna los fármacos con evidentes perfiles perjudiciales para la salud, y aplicar las sanciones y reparaciones a los afectados.
Además del fracaso regulatorio, que genera ineficiencias en el uso de recursos y daños evitables en la población, lo descrito por Prescrire cuestiona la eficacia del modelo vigente de I+D, pues los medicamentos verdaderamente innovadores y útiles que se desarrollan son pocos, mientras muchos pacientes siguen sin contar con terapias para abordar sus problemas de salud.
Referencias
Productos fraudulentos Sugar Control y Meta Fit podrían afectar órganos vitales y provocar efectos adversos severos
Portal WEB Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA)
Bogotá, 31 de julio del 2025
https://www.invima.gov.co/sala-de-prensa/productos-fraudulentos-sugar-control-y-meta-fit-podrian-afectar-organos-vitales-y
Tags: Alerta INVIMA por productos fraudulentos Sugar Control y Meta Fit, efectos adversos severos por consumo de sumplementos Sugar Control y Meta Fit, afectación de órganos vitales relacionado con consumo de productos fraudulentos Sugar Control y Meta Fit
COMUNICADO.
El Instituto Nacional de Vigilancia de Medicamentos y Alimentos – Invima, alerta a la ciudadanía sobre la comercialización ilegal de los productos Sugar Control y Meta Fit, que se están promocionando como suplementos dietarios sin contar con el respectivo registro sanitario. La venta de estos productos en Colombia es ilegal y representa un riesgo inminente para la salud de quienes los consumen.
Tras denuncias ciudadanas y procesos de verificación, el Invima confirmó que Sugar Control y Meta Fit no están amparados por registro sanitario alguno y, por tanto, no garantizan condiciones mínimas de calidad, seguridad y eficacia. Adicionalmente, presentan un etiquetado fraudulento y hacen uso de publicidad engañosa.
“Estos productos no cuentan con controles sobre su composición o trazabilidad, lo cual puede generar efectos adversos graves, desde trastornos cardiovascular hasta daños en órganos vitales”, explicó William Saza, coordinador de Farmacovigilancia del Invima, en relación con los riesgos asociados al consumo de los productos fraudulentos.
Entre los efectos adversos asociados se encuentran la elevación de la presión sanguínea, palpitaciones y posibles trastornos cardíacos, así como síntomas neurológicos como nerviosismo, ansiedad, temblores, insomnio y pesadillas. Además, su uso puede ocasionar retención de líquidos, edemas y daños potenciales en órganos vitales como el corazón, los riñones y el hígado.
Recomendaciones para la comunidad
Indicaciones para profesionales de la salud
Para establecimientos y autoridades sanitarias


