Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
Salud y Fármacos is an international non-profit organization that promotes access and the appropriate use of pharmaceuticals among the Spanish-speaking population.
La incierta prevención del cáncer de cuello de útero con la vacuna contra el virus del papiloma humano
Juan Gérvas [a]
Rev Port Clín Geral 2007;23:547-55
¿Cómo podemos asegurar la seguridad de la prescripción en los niños?
Traducido y resumido por Boletín Fármacos de: Sammons H y Conroy S, How do we ensure safe prescribing for children? Archives of Disease in Childhood 2008;93:98-99.
Productos Herbalife®: Publican series de casos de hepatitis tóxica y se evalúan los riesgos en España
Martín Cañás para Boletín Fármacos
Solicitud a la FDA para la prohibición de Ortho-Evra
Traducido por Boletín Fármacos de: Public Citizen, Petition to the FDA to Ban Ortho-Evra, 8 de mayo de 2008.
_______________________________________________________________________________
La incierta prevención del cáncer de cuello de útero con la vacuna contra el virus del papiloma humano
Juan Gérvas [a]
Rev Port Clín Geral 2007;23:547-55
Resumen
En 2007 se ha comercializado de la vacuna contra el virus del papiloma humano, con la que se propone vacunar a niñas de 11 y 12 años para la prevención primaria del cáncer de cuello de útero, dada la fuerte asociación entre el cáncer y algunos tipos oncogénicos del virus. La vacuna ha sido rápidamente incluida en los calendarios vacunales de la mayoría de los países desarrollados. En este texto se revisa el fundamento científico de dicha decisión. Son puntos claves: la ausencia de cambios en la epidemiología de la infección, la estabilidad o disminución de la incidencia y mortalidad del cáncer de cuello de útero, la falta de correlación entre respuesta inmunitaria serológica y la inmunidad natural, el impacto de la vacuna en la ecología del virus, las evaluaciones coste-efectividad que dependen de la duración desconocida de la inmunización, la dependencia excesiva de la investigación financiada por la industria farmacéutica, y la necesidad de mantener la citología de cribado. Se precisaría más tiempo e información antes de introducir la vacunación en el calendario vacunal.
Palabras clave: Vacunas, Virus del papiloma humano, Evaluación.
Once preguntas básicas (sin respuesta concluyente)
Con un ímpetu frenético, sin parangón en el campo vacunal, la vacuna contra el virus del papiloma humano se ha incluido en los calendarios vacunales de casi todos los países europeos, Alemania, Austria, Bélgica, Dinamarca, España, Grecia, Holanda, Italia, Luxemburgo, Reino Unido, Suecia y Suiza [1] y en otros desarrollados como Australia, Canadá y EE.UU.
¿La unanimidad en la decisión indica lógica y certeza científica? No. La prevención es campo aparte, como se deduce de otros casos; por ejemplo, respecto al cribado de la displasia del desarrollo de caderas en el recién nacido [2-4].
En el caso de la vacuna contra el virus del papiloma humano existen dudas razonables acerca de la racionalidad de la decisión de su inclusión en el calendario vacunal. Al menos hay once preguntas básicas sin respuesta concluyente, que hacen dudar de la oportunidad de la aprobación del nuevo calendario:
1. ¿Hay cambios recientes en lo que respecta a la infección por virus del papiloma humano? No. De hecho, desconocemos su historia natural. Es la enfermedad de transmisión sexual más frecuente y la más benigna (el 90% de las infecciones se curan espontáneamente) [5]. Seguimos sin saber porqué algunas infecciones son persistentes y cancerígenas (al cabo de 20-30 años provocan cáncer de cuello de útero).
2. ¿Hay cambios en los países desarrollados de la epidemiología del cáncer de cuello de útero que lo justifiquen? Por ejemplo, en España la incidencia se mantiene estable y baja, así como la mortalidad (respectivamente, de 7,11 y de 2,4 casos por 100.000 mujeres y año) [6]. En EE.UU. disminuye, y cada año hay unos 11.100 nuevos casos y unas 3.700 muertes por cáncer de cuello de útero [5].
3. La inmunidad natural ¿conlleva la presencia de anticuerpos en sangre? No. La cifra de anticuerpos en sangre es muy baja o inexistente (en la mitad de los casos) en las mujeres inmunes naturalmente. La infección no conlleva viremia (la replicación vírica se produce en la superficie epitelial, muy lejos de la células presentadoras de antígeno y de los macrófagos) [7]. Desconocemos en detalle la respuesta inmunológica normal, pero es muy efectiva. Además, no se ve afectada por la re-exposición debida a la actividad sexual continuada.
4. La vacuna, y re-vacuna, provoca la presencia en sangre de anticuerpos, en dosis de hasta veinte veces las máximas normales, pero ¿existe relación demostrada entre el nivel de anticuerpos y la eficacia de la vacuna? No. No hay correlación inmunológica demostrada. Ignoramos el mecanismo de acción de la vacuna. Se supone que los anticuerpos en sangre ayudan a eliminar los virus en la superficie epitelial, pero no sabemos cómo [5, 8]. La inmunidad natural es celular, no serológica.
5. Si la vacuna elimina los virus, ¿puede tener un doble efecto, uno beneficioso y otro perjudicial? Si. Por ejemplo, la vacuna disminuye las infecciones persistentes y las lesiones pre-malignas causadas por los virus contra los que se vacuna (beneficioso). Pero si eliminase otros virus del papiloma humano no sabríamos cómo valorarlo. Por ejemplo, la co-infección con los tipos 6 y 11 (bajo riesgo oncológico) disminuye naturalmente la posibilidad de ser infectado por el tipo 16 (alto riesgo oncológico) [9]. En general se acepta que la vacuna evita la presencia o actividad de los virus contra los que vacuna. Por ello cambia la “ecología” del cuello uterino y alrededores, y hay datos [10] que sugieren un efecto de “nicho vacío”, que permite la proliferación de otros virus de alto riesgo oncológico, o la transformación de los de bajo riesgo.
6. ¿Se ha demostrado su efectividad? No. No se tienen datos sobre su resultado en la práctica clínica diaria, ni siquiera ensayos clínicos con resultados en salud en las niñas en que se propone la vacunación. Se tienen datos de eficacia de casi el 100% (resultados de ensayos clínicos para los que cumplen todas las condiciones ideales, muy diferentes de la clínica diaria) para lesiones asociadas a los virus contra los que se vacuna, en mujeres de 16 a 26 años, generalmente blancas, sanas, de países desarrollados y educadas [10-14]. Cuando se tiene en cuenta “la intención de tratar” (se incluyen todos los pacientes participantes en los ensayos, aunque no hayan cumplido las condiciones ideales) la eficacia baja al 50% [10-14], y si se incluyen las lesiones no asociadas a los virus contra los que se vacuna, la eficacia baja hasta el 17% [11].
7. ¿Se sabe cuánto dura la inmunidad? No, no se sabe. Lo máximo demostrado son cinco años. Si la inmunidad decae, se podría precisar de una re-vacunación cada cierto tiempo. Además del gasto y complicaciones que ello implica, no sabemos si al ceder la inmunidad artificial se debilitaría la inmunidad natural y habría infecciones oncogénicas más graves y agresivas (algo parecido sucede con la vacunación contra la varicela) [7,12].
8. ¿Se ha determinado el coste-efectividad de la vacuna? Sí. Pero se asumen condiciones no demostradas. Especialmente respecto a la efectividad y respecto a la duración de la vacuna. De hecho, en condiciones muy probables, si la inmunidad provocada por la vacuna dura menos de treinta años, y si la efectividad es del 70%, en Canadá, el coste-efectividad es nulo. Es decir, habría que vacunar a infinitas niñas para evitar un caso de cáncer de cuello de útero [15].
9. ¿Sirve en mujeres que ya han iniciado la actividad sexual? No. Las mujeres se contagian al comienzo de la actividad sexual. La eficacia (ensayos clínicos, condiciones ideales) es muy baja en mujeres que ya han iniciado la actividad sexual, de alrededor del 17% [10,16]. Es una vacuna profiláctica (que evita el contagio), no terapéutica (que elimine el virus de las células epiteliales) [5,10].
10. ¿Hay ensayos clínicos y estudios independientes, no financiados o promovidos por la industria farmacéutica? No, o son irrelevantes. El grueso de la investigación sobre la vacuna contra el virus del papiloma humano ha dependido, depende y dependerá de la industria que fabrica dichas vacunas [10]. Se ignora porqué los gobiernos de los países desarrollados han renunciado a tener un papel activo en el conjunto de la salud sexual, y se reservan sólo el papel pasivo de “pagador” de la vacuna.
11. ¿Se precisa mantener el programa actual de detección precoz del cáncer de cuello de útero? Sí. Los actuales programas de cribaje con la citología (Papanicolau) tienen graves problemas de cobertura y fundamento científico, pero la vacuna no los evita, pues combate sólo dos de los quince virus oncogénicos. No sabemos en qué forma se modificará la sensibilidad y especificidad del cribaje [5,7].
Nota:
a. Juan Gérvas es Médico de Canencia de la Sierra, Garganta de los Montes y El Cuadrón (Madrid) España – Equipo CESCA, Madrid, España. Contacto: jgervasc@meditex.es
Bibliografía:
1. Vacuna.org. Calendario de vacunación. Europa. www.vacunas.org/index.php?option=com_content&task=view&id=2472&Itemid=336. Consultado el 13 de octubre de 2007.
2. Gervas J, Pérez Fernández M, González de Dios J. Problemas prácticos y éticos de la prevención secundaria. A propósito de dos ejemplos en pediatría. Rev Esp Salud Pública 2007;81:345-52.
3. Starfield B, Hyde J, Gérvas J, Heath I. The concept of prevention: a good idea gone astray? J Epidemiol Community Health 2008 [in press].
4. Gérvas J, Starfield B, Heath I. Caution in clinical prevention. Lancet 2008 [in press].
5. Markowitz LE, Dunne EF, Saraiya M, Lawson HW, Chessm H, Unger E for the ACIP. Quatrivalent human papilloma virus vaccine. MMWR 2007;56(RR02): 1-24.
6. Galceran J, Marcos R, Izquierdo A, Borrás J. Carcinoma invasor y lesiones premalignas del cuello uterino en los registros poblacionales: utilidad y limitaciones. En: Sanjosé S, García A (coordinadoras). Madrid: Sociedad Española de Epidemiología (4ª Monografía); 2006. p. 15-29.
7. Navarro JA, Bernal PJ, Pérez JJ. Interrogantes en la introducción de la vacuna frente al virus del papiloma humano en los calendarios sistemáticos. Med Clín (Barc) 2007;129:55-60.
8. EMEA. EPARS for authorised medicinal products. Gardasil. www.emea.europa.eu/humandocs/Humans/EPAR/gardasil/gardasil.htm. Consultado el 13 de octubre de 2007.
9. Hughes JP, Garnett GP, Koutsky L. The theoretical population-level impact of a prophylactic human papilloma virus vaccine. Epidemiol 2002;13: 361-9.
10. Sawaya GF, Smith K. HPV vaccination. More answers, more questions. N Engl J Med 2007;356:1991-3.
11. Kahn JA, Burk RD. Papillomavirus vaccines in perspective. Lancet 2007;369:2135-7.
12. Lippman A, Melnychuk R, Shimmin C, Boscope M. Human papillovirus, vaccines and women’s health: questions and cautions. CMAJ 2007;177:484-7.
13. Joura EA, Leodoter S, Hernández-Ávila M, Wheeler CM, Pérez G, Loutsky LA et al. Efficacy or quadrivalent prophylactic human papillomavirus (types 6, 11,16, and 18) L1 virus-like-particle vaccine against high-grade vulvar, and vaginal lesions: a combined analysis of three randomised clinical trials. Lancet 2007;369:1693-702.
14. Rambout L, Hopkins L, Hutton B, Fergusson D. Prophylactic vaccination against human papillomavirus infection and disease in women: a systematic review of randomized controlled trials. CMAJ 2007;177:469-79.
15. Brisson M, Velde N, Wals P, Boily MC. Estimating the number needed to vaccinate to prevent diseases and death related to human papillomavirus infection. CMAJ 2007;175:464-8.
16. Future II Study Group. Quatrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007:356:1915-27.
¿Cómo podemos asegurar la seguridad de la prescripción en los niños?
Traducido y resumido por Boletín Fármacos de: Sammons H y Conroy S, How do we ensure safe prescribing for children? Archives of Disease in Childhood 2008;93:98-99.
Entre enero 2005 y junio 2006 se reportaron a la UK National Patient Safety Agency casi 10.000 incidentes de seguridad de medicamentos relacionados a la prescripción y más del 80% ocurrieron en los hospitales [1].
Más del 10% de todos los incidentes en donde se pudo establecer la edad involucraron a niños de hasta 4 años de edad, lo que representa un porcentaje superior a los días-cama de hospitalización de este grupo de edad. Es probable que estas cifras estén subestimadas porque provienen de reportes voluntarios; sin embargo, la renuencia a reportar errores en el sistema nacional de salud esta gradualmente cambiando ya que se reconoce cada vez más que los culpables no son los individuos sino los sistemas.
La competencia de los profesionales médicos para prescribir ha sido objeto de reciente debate en la prensa médica. Aronson et al. [2] resaltaron el hecho de que los estudiantes de medicina y los médicos jóvenes podrían estar poco preparados para prescribir medicamentos cuando se gradúan. La discusión subsiguiente llevó a que el comité de educación del General Medical Council (GMC) del Reino Unido hiciera una evaluación de la enseñanza y de la prescripción. Esto ha promovido planes para investigar la prevalencia y las causas de los errores de prescripción y para identificar recomendaciones de intervenciones educativas o éticas que contribuyan a reducirlos (ver www.gmc-uk.org/education/documents/pap_prescribingITT_v1.0.pdf )
Se ha descrito un currículum que será el mismo para todas las escuelas de medicina del Reino Unido y que servirá para capacitar sobre la prescripción segura y efectiva [3]. Este currículum menciona a los niños en dos secciones:
a) La prescripción en pacientes con requisitos especiales debe ser uno de los ejes centrales del conocimiento y de la comprensión porque estos grupos tienen una fisiología alterada, y el manejo farmacocinético y la respuesta farmacodinámica pueden ser diferentes.
b) El núcleo de habilidades deben incluir prescripción de medicamentos a grupos especiales.
Sin embargo, las secciones no incluyen información más detallada.
Los niños no son adultos pequeños en lo que respecta al desarrollo o prescripción de medicamentos. La disponibilidad de información para respaldar la prescripción pediátrica ha mejorado en los últimos años, se ha pasado de utilizar una plétora de formularios pediátricos y guías de dosis basadas en la práctica local a tener un Formulario Nacional Británico para Niños (BNF-C por sus singlas en inglés) que contiene una base de datos nacionales basados en la evidencia y revisados por pares. Sin embargo, los errores en prescripción continúan tanto en niños como en adultos. Además, hemos incrementado el número de prescriptores no-médicos, como enfermeras y farmacéuticos, que están prescribiendo a niños.
¿Por qué son los niños diferentes?
Los niños requieren dosis de medicamentos calculadas en forma individual, teniendo en cuenta la edad gestacional y postnatal, y el peso o superficie corporal. Comparado a la situación en adultos, las dosis “estándar” raramente existen. Para seleccionar el medicamento y la dosis se debe tener en cuenta los cambios dinámicos que experimentan los niños en la farmacocinética y la madurez farmacodinámica, y la posible toxicidad medicamentosa, la cual puede ser muy diferente a la de los pacientes adultos. La necesidad de utilizar medicamentos no aprobados o llamados “off label” para muchos niños tanto cuando están hospitalizados como cuando se trata de pacientes ambulatorios señala que no se han comercializado los productos adecuados y que la información para la prescripción no siempre está disponible. Los niños están en mayor riesgo de ser victimas de errores de prescripción que los adultos y tienen menores reservas internas para contrarrestar estos efectos cuando ocurren [4].
Facultad de Medicina: Enseñando prescripción y terapéutica
Se tiene la percepción de que se ha reducido la enseñanza de terapéutica clínica y prescripción en las facultades de medicina. Tradicionalmente las facultades de medicina enseñaban “ciencias básicas” como base fundamental para la clínica pero ahora muchos usan un sistema curricular de aprendizaje basado en problemas. El GMC ha dado los lineamientos sobre los objetivos claves de aprendizaje que tienen que ver con el uso de medicamentos en el manejo de enfermedades (www.gmcuk.org/education/undergraduate/undergraduate_policy/tomorrows_doctors.asp). Esto requiere que los estudiantes sean capaces de calcular las dosis y registrar los resultados de modo muy preciso; y de escribir prescripciones seguras para los distintos tipos de medicamentos. Los estudiantes de medicina, sin embargo, parecen sentirse menos confiados en sus habilidades para prescribir al momento de graduarse de la universidad.
En una encuesta a médicos F1 (médicos que rotan por distintas áreas como medicina interna, cirugía), el 68% no estuvo de acuerdo con la afirmación: “Fui entrenado adecuadamente para prescribir en el momento de mi graduación” [5]. Aunque se tiene la percepción de que los errores de prescripción podrían reflejar la falta de capacitación adecuada durante la formación de los médicos, hay poca investigación al respecto [3].
Enseñando a los médicos jóvenes
Muchos departamentos de pediatría en el Reino Unido desarrollan sus propias sesiones de enseñanza para la prescripción a niños. Se ha dado poca difusión a los formatos o contenidos, y la mayoría de los centros trabaja independientemente sin compartir recursos ni experiencias. No hay recomendaciones sobre la enseñanza de los nuevos médicos o para el entrenamiento de los pediatras. En nuestra organización, desarrollamos una sesión de enseñanza de una hora para los senior house officers (SHO, por sus siglas en inglés. Son médicos que están en capacitación a nivel de especialidad médica) durante su primer mes de rotación en pediatría y una sesión de dos horas para todos los médicos F2. Estas sesiones incluyeron una presentación sobre las diferencias entre la prescripción a pacientes pediátricos y a adultos, mencionaron la importancia de hacer investigación en medicamentos, resaltaron la importancia de los riesgos de errores de medicación en pacientes pediátricos y se presentaron ejemplos de errores comunes.
Se presentaron diferentes escenarios y se solicitó a los médicos que prescribieran, y luego hubo una discusión. No hay una evaluación formal de la prescripción individual de los médicos. En el nivel de hospitalización, los farmacéuticos y enfermeras juegan un rol importante en la detección y rectificación de errores, y proveen recomendaciones y respaldo al staff médico en lo que hace referencia a la prescripción. Un estudio de seis semanas de duración que se realizó en nuestra institución identificó 139 intervenciones clínicas hechas por farmacéuticos y enfermeras para corregir o clarificar prescripciones [6].
Evaluación de la competencia: Más trabajo
Mientras el proyecto de investigación de GMC dirigido a establecer la prevalencia y las causas de los errores de prescripción entre los nuevos médicos fue bien recibido, los retos para los pediatras son específicamente distintos a los que ocurren en la prescripción a adultos. Sería prudente investigar las medidas que se están tomando para capacitar en la prescripción en pediatría en el Reino Unido y las evaluaciones de competencia que se estén haciendo. Es esencial que nuestras organizaciones nacionales como Royal College of Paediatrics and Child Health (RCPCH) y Neonatal and Paediatric Pharmacists Group se involucren en el desarrollo de estándares nacionales.
Algunos esperan que la prescripción electrónica sea la respuesta a estos problemas y se ha sugerido que reduce las tasas de errores de prescripción; sin embargo, su introducción en el Reino Unido será todo un reto y más aun para los sistemas de prescripción en pacientes pediátricos, que requieren un sistema de apoyo más complejo que el de los adultos, y es improbable que en los próximos años esté ampliamente disponible para los neonatos y niños.
Conclusión
La seguridad y bienestar del paciente necesita ser la primera preocupación de cualquier profesional de la salud, sin importar la edad del paciente.
La prescripción en pediatría, sin embargo, tiene sus propios retos específicos. Los médicos que trabajen en pediatría y aquellos en capacitación necesitan por tanto tener acceso a formación y supervisión continuada. En el Reino Unido, no tenemos entrenamiento formal para la prescripción, ni una herramienta validada para evaluar la competencia en la prescripción para cualquier grupo etáreo. Estas herramientas son esenciales para evitar los errores que ocurren regularmente. Si vamos a llevar a cabo la evaluación de la competencia, necesitamos tener mecanismos para solucionar los problemas de prescripción que se identifiquen y para capacitar a los prescriptores. Además, se necesitarán mecanismos para hacer nuevas evaluaciones en el futuro y los recursos para solucionar los problemas que se descubran. Estos serán los retos para los pediatras, farmacéuticos y otros profesionales no-médicos que prescriben.
Ejemplos de medicamentos con posible consecuencias serias si se prescriben incorrectamente:
– Aminoglucósidos
– Anticoagulantes
– Drogas citotóxicas
– Digoxina
– Insulina
– Opioides
– Fenitoína
– Cloruro de potasio
Referencias:
1. National Patient Safety Agency. Safety in doses: improving the use of medicines in the NHS. London: NPSA, 2007.
2. Aronson JK, Henderson G, Webb DJ, et al. A prescription for better prescribing. BMJ 2006;333:459-60.
3. Maxwell S, Walley T. Teaching safe and effective prescribing in UK medical schools: a core curriculum for tomorrow’s doctors. Br J Clin Pharmacol 2003;55:496–53.
4. Kaushal R, Bates DW, Landrigan C, et al. Medication errors and adverse drug events in pediatric inpatients. JAMA 2001;285:2114–20.
5. Han WH, Maxwell SR. Are medical students adequately trained to prescribe at the point of graduation? Views of first year foundation doctors. Scot Med J 2006;51:27–32.
6. Conroy S, Appleby K, Bostock D, et al. Medication errors in a children’s hospital. Paediatr Perinat Drug Ther 2007;8:18–25.
Productos Herbalife®: Publican series de casos de hepatitis tóxica y se evalúan los riesgos en España
Martín Cañás para Boletín Fármacos
Los suplementos dietarios son productos destinados a incrementar la ingesta dietaria habitual. Suplementan la incorporación de nutrientes en la dieta de las personas sanas que, no encontrándose en condiciones patológicas, presenten necesidades básicas dietarias no satisfechas o mayores a las habituales. Como todos los alimentos, se venden libremente. Pueden contener en su composición, en forma simple o combinada, los siguientes ingredientes: péptidos, proteínas, lípidos, lípidos de origen marino, aminoácidos, glúcidos o carbohidratos, vitaminas, minerales, fibras dietarios y hierbas. Debe quedar claro que, en condiciones normales, la dieta debe proveer todos los nutrientes necesarios para el mantenimiento de las funciones del organismo. Por lo tanto, un suplemento dietario sólo debería consumirse cuando, por un estado fisiológico particular, el individuo necesite un suministro extra de un nutriente determinado, previa consulta al médico [1].
Los productos Herbalife están calificados legalmente en España y toda la Unión Europea y en casi todos los países, como alimentos (bien Alimentos Destinados a Regímenes Especiales o bien Complementos de la Dieta, dependiendo de su composición), alegando generalmente su utilidad para perder peso o mejorar el bienestar de las personas que los utilizan [2]. Se comercializan fundamentalmente a través de venta directa mediante los propios distribuidores de la compañía Herbalife y a través de Internet. Se encuentran disponibles diversos productos bajo diferentes denominaciones, cuya composición es variable, pero esencialmente a base de plantas, asociadas en algunos casos a vitaminas, minerales u otros nutrientes [2].
A fines de julio de 2007 se difundieron en forma anticipada por la revista Journal of Hepatology, la publicación de dos series de casos que informaron sobre una posible relación entre lesiones hepáticas y los productos Herbalife®. Ambos trabajos señalan que los productos herbolarios son muy populares y que se los considera seguros porque son supuestamente “naturales”. Sin embargo, en sus series presentan un total de 22 casos de hepatitis tóxica asociada al consumo de productos Herbalife [3,4].
Según relata el trabajo de Elinav y cols., cuatro casos iniciales de hepatitis aguda asociada al consumo de productos Herbalife motivaron una investigación del Ministerio de Salud de Israel en todos los hospitales de ese país. Como parte del estudio se investigaron 12 pacientes con injuria hepática aguda idiopática, asociada al consumo de Herbalife. Once pacientes eran mujeres, de 49 años promedio (+/- 13). Una paciente tenía cirrosis biliar primaria y otra antecedentes de hepatitis B. El tiempo de consumo promedio, antes de padecer la injuria hepática fue de 11,9 meses (+/-11,1). Las biopsias hepáticas mostraron hepatitis activa, inflamación portal con eosinófilos, reacción ductular e inflamación parenquimatosa con actuación pericentral. Un paciente desarrolló hepatitis subfulminante y dos episodios fulminantes de insuficiencia hepática. La hepatitis se resolvió favorablemente en 11 pacientes, mientras que el restante falleció debido a complicaciones del transplante hepático. Luego de la normalización enzimática, tres pacientes volvieron a consumir los productos presentando un segundo episodio de hepatitis, que en dos casos se resolvió suspendiendo nuevamente el consumo de Herbalife y el otro estaba aun en tratamiento al momento de la publicación del estudio.
De los 12 pacientes, nueve consumían productos para reducir de peso y tres para “sentirse bien”. Dos de ellos aumentaron el consumo de productos Herbalife al consultar a su distribuidor por los síntomas que presentaban.
Los autores concluyen que se identificó la asociación entre la ingesta de productos Herbalife y hepatitis aguda. Hacen un llamamiento para que se efectúe un análisis de estos productos y poder evaluar su posible hepatotoxicidad. Hasta entonces, recomiendan que los consumidores tengan precaución, especialmente aquellos que padecen enfermedad hepática subyacente [3].
En el segundo trabajo, Schoepfer y cols. presentan 10 casos de hepatitis tóxica asociados al consumo de productos Herbalife. El estudio se llevó a cabo en Suiza para determinar la prevalencia y la evolución de la hepatotoxicidad asociada al consumo de Herbalife, para lo cual se envió un cuestionario a todos los hospitales públicos suizos. Los casos notificados fueron sometidos a la evaluación de la causalidad utilizando los criterios de CIOMS. Como resultado se recuperaron doce casos de hepatitis tóxica en los cuales estaban implicados los preparados Herbalife (1998-2004), 10 de ellos suficientemente documentados como para permitir el análisis de la causalidad. La mediana de edad de los pacientes fue de 51 años (rango 30-69) y la latencia de inicio fue de 5 meses (desde 0,5 hasta 144). La biopsia hepática (7/10) mostró necrosis hepática, infiltración linfocítica / eosinofílica y coléstasis marcadas en cinco pacientes. Un paciente con insuficiencia hepática fulminante fue exitosamente trasplantado; el explante mostró hepatitis con células gigantes. En un caso se observó síndrome de obstrucción sinusoidal. Tres pacientes sin biopsia hepática presentaron lesión hepática con patrón hepatocelular (2) o mixto (1). La evaluación de la causalidad de las RAMs, fue clasificada como de certeza en dos casos, probable en siete y posible en un caso. Los autores concluyen que la hepatitis tóxica asociada al consumo de productos Herbalife puede ser grave. Asimismo afirman que sería deseable [conocer los componentes de la fórmula y que los organismos reguladores tengan un papel proactivo en el uso y autorización de estos productos [4].
Las advertencias en España
En abril de 2008, el Ministerio de Sanidad y Consumo difundió un comunicado en el que informaba el comienzo de una investigación para conocer la causa de nueve casos de toxicidad hepática aparecidos entre 2003 y 2007, y su posible relación con la ingesta de productos dietéticos de la empresa Herbalife [2].
La comunicación conjunta de la Agencia Española de Seguridad Alimentaria y Nutrición (AESAN) y la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), informa que el Sistema Español de Farmacovigilancia (SEFV) recibió nueve casos de alteraciones hepáticas (cinco por tarjeta amarilla y cuatro procedentes del Registro de Hepatopatías de la Universidad de Málaga), producidos entre 2003 y 2007, y en los que diversos productos Herbalife se consideraron sospechosos de producir alteraciones de las enzimas hepáticas y lesiones hepáticas graves [2].
A través de la consulta con los responsables de farmacovigilancia de los demás Estados Miembros de la Unión Europea (UE), se obtuvo información sobre nueve casos más de hepatotoxicidad en la UE, producidos entre 1992 y 2006, y de otros 6 en Islandia. La AEMPS también informa de las series de casos de Suiza y de Israel mencionadas arriba. La AEMPS señala que la mayoría de los pacientes se recuperaron y normalizaron su función hepática tras dejar de utilizar el producto.
El informe de la AEMPS señala la dificultad de establecer la relación de causalidad en estos casos, y que la fortaleza de esta asociación fue variable entre unos casos y otros. Esto en parte se debió a que en algunos de ellos existían medicamentos concomitantes sospechosos de poder producir alteraciones hepáticas y también a que se detectaron conductas de abuso con ingestión de grandes cantidades, ingestión de diversos productos simultáneamente o mantenimiento del tratamiento durante largo tiempo.
Sin embargo, la posible asociación de estas alteraciones con el consumo de productos Herbalife, estaría apoyada por la reaparición de la alteración hepática observada en algunos casos cuando se produjo una reexposición, después de la suspensión del tratamiento [2,5].
La Agencia señala que en base a la información disponible actualmente no es posible concluir si la aparición de alteraciones hepáticas se asocia a productos concretos, a un componente determinado o a otras causas presentes en estos productos, dado que los pacientes utilizaron distintos y variados productos de Herbalife.
El Comité de Seguridad de Medicamentos de Uso Humano (CSMH) de la AEMPS concluyó que la información disponible sugiere una relación causal entre el consumo de productos Herbalife y las alteraciones hepáticas registradas. Además, el CSMH indicó que, aunque la frecuencia se desconoce por disponer únicamente de información basada en notificaciones espontáneas, esta asociación puede constituir un problema de salud pública [2]
Repercusiones del informe de la AEMPS, la voz de la empresa y de los consumidores
En respuesta a las recomendaciones del Ministerio de Sanidad, la empresa Herbalife difundió un comunicado el 22 de abril. La empresa señala que Herbalife planea someter la sección final de su detallado dossier de productos al Ministerio de Sanidad español, que incluirá información adicional relacionada con la seguridad de sus productos. Según indica la empresa, esto es parte del diálogo que vienen teniendo desde hace tiempo ambas partes y Herbalife continuará cooperando totalmente con las autoridades españolas [6].
Un asesor externo de Herbalife en Argentina, sostuvo que los consumidores de los productos “tienden a ser personas que padecen sobrepeso u obesidad, lo que ya de por sí aumenta las probabilidad de sufrir problemas hepáticos” [7].
Este asesor señaló que no hay estudios científicos que demuestren la toxicidad de los “alimentos” de Herbalife, aunque tampoco hay estudios integrales que demuestren que no representan riesgos para la salud. También explicó que, por no tratarse de un medicamento, “ningún país del mundo” requiere análisis de este tipo para lanzar los productos al mercado, aunque la empresa lleva a cabo estudios clínicos “en Alemania y Brasil”. Claus Donath, director general de Herbalife Argentina, afirmó al periodismo que “este es un tema que viene desde hace años y nunca se pudo probar” y atribuyó las denuncias a los competidores de la empresa [7].
Por su parte, la Confederación de Consumidores y Usuarios (CECU) de España pidió a Sanidad que se pronunciase claramente sobre los posibles efectos sobre la salud de los productos Herbalife. En un comunicado, ha asegurado que la administración debe decir si se puede o no seguir consumiendo estos complementos alimentarios hasta que se confirmen o rechacen los resultados de los análisis y pruebas que se están realizando [8].
Consideraciones generales sobre los productos dietarios y medicina alternativa y complementaria
En un editorial acompañante a los trabajos de Suiza e Israel, Sitckel [5] reflexiona sobre cómo las medicinas complementarias y alternativas (CAM, por sus siglas en inglés) están experimentando un auge sorprendente debido al creciente interés de la opinión pública en la profilaxis de enfermedades, nutrición y en las mejoras para la salud y el bienestar. Remarca que una contribución a este proceso incluye a los suplementos nutricionales como vitaminas, antioxidantes y hierbas, fórmulas dietéticas para disminuir de peso, y preparados -mal definidos- como para mantener el cuerpo en “forma”.
La mayoría de los individuos que utilizan estos productos deberían considerarse “clientes” en lugar de “pacientes”, ya que no pretenden específicamente tratar ninguna enfermedad, sino mejorar su salud en general. La mayor parte de la utilización de hierbas y suplementos alimenticios ocurre como automedicación, a expensas de un gasto personal considerable y sin consulta previa y conocimiento de sus médicos. Menciona los datos de una encuesta nacional de EE.UU. que mostró una prevalencia entre el 37,5% y el 67%, y un aumento constante, en el uso de hierbas medicinales y suplementos nutricionales.
Aunque para Stickel se podría dar la bienvenida a este creciente desarrollo, ya que refleja en el público una mayor conciencia acerca de la prevención de las enfermedades y la salud, aun si los reales beneficios de estas sustancias son pequeños para los consumidores, reconoce que existen varios problemas, entre ellos:
a- Se han acumulado informes sobre reacciones adversas, en particular de lesiones hepáticas, por lo que debería considerarse que este tipo de productos no siempre son tan inofensivos como se piensa.
b- En la mayor parte de los países occidentales, incluidos la UE y EE.UU., los suplementos nutricionales y preparados a base de hierbas, están exentos de las estrictas regulaciones para conceder la autorización de comercialización, que es obligatoria para los medicamentos o fármacos sintéticos antes de su ingreso al mercado. Los preparados de CAM son tratados como alimentos, para los cuales no se necesita una preautorización para su comercialización. La responsabilidad sigue siendo totalmente de la empresa y no tienen que aportarse pruebas de seguridad, y mucho menos de eficacia.
c- La composición de la mayoría de los productos no está suficientemente clara, frecuentemente no incluida en el etiquetado, altamente variable y no existen claros efectos beneficiosos para los consumidores.
d- Las empresas afirman que las supuestas ventajas para la salud sólo se perciben a través de su uso regular y a largo plazo, aunque estas afirmaciones no están respaldadas por pruebas clínicas. Por lo tanto, los frecuentemente elevados costos de estos productos contrastan con la ausencia de pruebas sobre sus beneficios.
e- En general los profesionales de la salud conocen poco acerca de los suplementos dietarios y productos herbarios, y pocos los perciben como fuente probable de riesgos para la salud
Por otra parte, Stickel recuerda lo que sugieren los autores de la serie suiza: la amenaza a la salud pública de los productos Herbalife no debe exagerarse, sobre todo cuando se compara con las tasas de incidencia de reacciones adversas hepáticas de otros productos farmacéuticos de venta libre como antiinflamatorios no esteroideos (AINEs) [5].
¿Exceso de plomo?
Un mes después de la comunicación española, el Fraud Discovery Institute (Instituto de Hallazgo de Fraude) colocó una carta en su portal de internet atribuida a Christopher Grell, abogado de Oakland, California, especializado en responsabilidad por fabricación de productos, donde señalaba que las dosis diarias recomendadas de seis productos de Herbalife contenían niveles de plomo peligrosos y que superan la legislación californiana al respecto [9].
La legislación obliga a los empresarios a advertir a los consumidores sobre si sus productos contienen químicos que causan cáncer o toxicidad reproductiva.
La carta colgada en la web del Instituto [10] instaba a California a ordenar a Herbalife colocar “advertencias claras y razonables sobre estos productos”, para que el consumidor esté informado de que contienen químicos que se sabe que causan daños para el desarrollo.
Los seis productos mencionados en la misiva son ShapeWorks Protein Drink Mix, Healthy Meal Nutritional Shake Mix, las tabletas Tang Kuei Plus, el té instantáneo Thermojetics Nature’s Raw Guarana, ShapeWorks Cell Activator y Multivitamin Complex.
La web tiene también un supuesto recibo de un laboratorio de la empresa Anaheim, con sede en Brea, California, que habría analizado los seis productos. Analytical Laboratories no contestó a las llamadas ni a un correo electrónico para comentar las pruebas.
Herbalife cuestionó esta afirmación, diciendo que sus productos cumplen con los requisitos reguladores de todos los mercados en los que están presentes.
“La FDA (Administración de Alimentos y Fármacos de Estados Unidos) no ha establecido un límite general sobre el plomo en los alimentos, pero nosotros estamos dentro de sus directrices recomendadas”, dijo el portavoz de Herbalife, George Fischer, en una entrevista telefónica [9].
En un comunicado de prensa Herbalife señalaba también que “Es una irresponsabilidad vincular la Proposición 65, la cual tiene que ver con cuestiones de divulgación, con la seguridad de nuestros productos. La Proposición 65 es una ley de divulgación y etiquetado que requiere, bajo ciertas circunstancias, la divulgación de la presencia de cualquiera de aproximadamente 800 químicos señalados (incluyendo el plomo).” Y continúa diciendo que “Nuestros productos incluyen ingredientes naturales y la existencia natural de niveles de residuo de plomo está presente en prácticamente todos los ingredientes naturales. Esta no es una cuestión sobre la contaminación como resultado del proceso de fabricación o como resultado del tratamiento inseguro.” “Nosotros cumplimos con todos los procesos escritos de control de seguridad y calidad. Respaldamos la seguridad de nuestros ingredientes y productos”, concluye [11].
Recomendaciones a los profesionales sanitarios de la AESAN y la AEMPS [2]
– Realizar una anamnésis detallada en aquellos pacientes con alteraciones hepáticas al objeto de obtener información sobre el uso de productos Herbalife o cualquier otro producto no medicamentoso que el paciente haya utilizado, bien como complemento alimenticio o como producto a base de plantas medicinales con cualquier otro fin. Considerar la posibilidad de interacción con alimento o medicamento.
– Notificar al Sistema de Farmacovigilancia correspondiente cuando se sospeche una reacción adversa.
– Pedir el envase del producto al paciente para su identificación y posterior análisis, si procede, el cual debe remitirse a las autoridades sanitarias [2].
En síntesis, como se señaló anteriormente, muchas veces la información sobre este tipo de productos es presentada de forma tal que alienta expectativas poco realistas sobre los efectos beneficiosos, en algunos pocos caso no están exentos de riesgos y los profesionales de salud en general tienen nociones vagas sobre toda esta gama de productos [5].
Es necesario recordar a los pacientes que están por empezar -o ya comenzaron- dietas para adelgazar, que es importante que consulten con profesionales de salud para establecer una planificación para garantizar una nutrición adecuada y comidas bien balanceadas, y que los suplementos nunca deben ser tomados como la única fuente de alimentación. Si bien la mayoría de los suplementos dietarios son de venta libre, es importante consultar siempre al médico o farmacéutico ya que lo “natural” no siempre está exento de riesgos.
Nota de los editores:
– Recomendamos ver: “Productos para adelgazar: Se prohíbe la comercialización de Herbalife en Paraguay” en la Sección Advierten del Boletín Fármacos 2005;8(2).
Referencias:
1. ANMAT, Verdades y mentiras de los suplementos dietarios. Disponible en: www.anmat.gov.ar/consumidores/alimentos/Suplementos_Dietarios_Verdades_Mentiras.pdf
2. AEMPS / AESAN, Posibles alteraciones hepáticas asociadas al consumo de productos HERBALIFE® Comunicación sobre riesgos de medicamentos para profesionales sanitarios, Ref: 2008/07, 21 de abril de 2008. Disponible en: www.agemed.es/actividad/alertas/usoHumano/seguridad/docs/NI-HERBALIFE-abril08.pdf
3. Elinav E et al., Association between consumption of Herbalife ((R)) nutritional supplements and acute hepatotoxicity, J Hepatol 2007;47(4):514-20.
4. Schoepfer AM et al., Herbal does not mean innocuous: Ten cases of severe hepatotoxicity associated with dietary supplements from Herbalife((R)) products, J Hepatol 2007;47(4):521-6.
5. Stickel F, Slimming at all costs: Herbalife® -induced liver injury, J Hepatol 2007;47:444-46.
6. Herbalife, Comunicado de Herbalife en respuesta a las recomendaciones del Ministerio de Sanidad, 22 de abril de 2008. Disponible en: www.herbalifeww.com/es/pdf/News_More_Info_Items/Comunicado_Herbalife.pdf
7. Investigan si son tóxicos los productos de Herbalife, Diario Perfil, 21 de abril de 2008.
8. Sanidad investiga varios casos de toxicidad y su posible relación con productos Herbalife. El Mundo (España), 21 de abril de 2008.
9. Geller M, Grupo de EE.UU. dice productos de Herbalife contienen mucho plomo, Reuters, 20 de mayo de 2008.
10. Grell C, Lead findings Attorneys At Law specializing in ephedra litigation, asbestos, dietary supplements, and PPA, 16 de mayo de 2008. Disponible en: www.frauddiscovery.net/herbalife2/Lead_findings.pdf
11. Declaración de Herbalife, PRNewswire-HISPANIC PR WIRE, 19 de mayo de 2008. Disponible en: www.hispanicprwire.com/generarnews.php?l=es&id=11539&cha=0
Solicitud a la FDA para la prohibición de Ortho-Evra
Traducido por Boletín Fármacos de: Public Citizen, Petition to the FDA to Ban Ortho-Evra, 8 de mayo de 2008.
Andrew von Eschenbach, M.D.
Comisario de la Agencia de Alimentos y Fármacos de EE.UU.
Food and Drug Administration
5600 Fishers Lane
Rockville, Maryland 20857 EE. UU.
Estimado Dr. von Eschenbach:
Por la presente, Public Citizen, que representa a más de 80.000 consumidores de todo el país, solicita a la Agencia de Alimentos y Fármacos (FDA), de acuerdo con la Ley Federal de Alimentos, Fármacos y Cosméticos 21 U.S.C. Sección 355(e)(3) y 21 C.F.R. 10.30, la prohibición del parche transdérmico anticonceptivo Ortho-Evra (etinil estradiol/norelgestromin, Jonson & Johnson) y la retirada comercial del dispositivo en un plazo de seis meses.
En comparación con anticonceptivos orales estándar de estrógenos y progestina de 35 microgramos (mcg), Ortho-Evra presenta:
– Una exposición media a los estrógenos el 60% más elevada;
– Una mayor variabilidad en los niveles de estrógenos;
– Posiblemente el doble de riesgo de trombosis venosa (coágulos de sangre dolorosos de las extremidades inferiores que pueden desplazarse hasta los pulmones por el torrente sanguíneo y causar la muerte);
– Un aumento del riesgo de efectos secundarios, como molestias en las mamas, dolor menstrual severo, náuseas y vómitos;
– Un incremento del 50% de probabilidades de interrupción del tratamiento;
– Sin mejoría significativa en la efectividad como contraceptivo.
En los últimos años, millones de mujeres y sus facultativos han descubierto que Ortho-Evra es un producto menos atractivo que lo anunciado. La demanda de los parches ha experimentado un drástico descenso, desde más 9,9 millones de recetas en el año 2004 a 2,7 millones de recetas en 2007 (disminución del 73%). Sin embargo, en el año 2007 Ortho-Evra permanecía entre los 200 de fármacos de marca comercial de mayores prescrpciones y ventas en EE.UU. [1].
Los litigios en curso en nombre de las mujeres afectadas o fallecidas por el uso de Ortho-Evra han permitido que se hagan públicos los resultados de dos estudios no publicados previamente que muestran que Johnson & Johnson conocía los niveles potencialmente altos de estrógenos del parche, así como la mayor variabilidad de la administración de estrógenos antes de su aprobación por FDA en noviembre de 2001 [2]. El prospecto del producto nunca ha mencionado el aumento de probabilidades de efectos secundarios con el parche. Las mujeres merecen un nivel de riesgo similar al obtenido con los anticonceptivos orales combinados habituales (la “píldora”), especialmente cuando la eficacia contraceptiva del parche transdérmico no es superior.
Introducción
Ortho-Evra es un parche que contiene 0,75mg de etinil estradiol (EE, un estrógeno) y 6mg de norelgestromin (NGMN, una progestina). Cada parche está diseñado para aplicarse en la piel durante siete días consecutivos antes de su retirada. Cada ciclo consta de tres parches para un total de tres semanas de tratamiento más una semana de descanso. Según el prospecto, el parche puede colocarse en el abdomen, las nalgas, la parte superior externa de los brazos o la parte superior del torso. Johnson & Johnson consideraba que su parche presentaba dos ventajas principales en comparación con los anticonceptivos orales: (1) nivel estable de hormonas en sangre debido a la dosis semanal y a que se evitaba la eliminación inicial de las hormonas a través de su metabolismo normal por el tracto gastrointestinal y/o el hígado, y (2) mejoras del cumplimiento debido a la aplicación cutánea y al régimen semanal en sustitución del régimen de dosificación diario.
El análisis de esta petición se centra en la seguridad y la eficacia de la elevada biodisponibilidad de estrógenos en el caso de Ortho-Evra en comparación con la píldora de EE de 35mcg. Sin embargo, la comparación con el tratamiento oral resulta complicado debido a diferencias en los perfiles farmacocinéticos (FC). Por ejemplo, la ingesta de un anticonceptivo oral produce un pico pronunciado de estrógenos en la sangre antes de su metabolización durante 24 horas y la ingesta de la siguiente dosis. Por el contrario, la aplicación del parche resulta en un incremento gradual del nivel de estrógenos que alcanza la meseta a los tres días y que permanece relativamente constante durante la aplicación de los parches. Además, el estado estacionario, o el estado en el que los niveles en sangre de la hormona son estables, porque la tasa de excreción coincide con la tase de ingesta, se alcanza en diferentes momentos; alrededor del tercer día después de haber comenzado la terapia oral y en la segunda semana de la aplicación del parche.
La biodisponibilidad describe la cantidad de fármaco administrado que se absorbe en el torrente sanguíneo y que, por tanto, se encuentra disponible en el sitio de acción. Normalmente se determina con una combinación con parámetros farmacocinéticas (FC), incluido el máximo nivel de concentración química en la sangre (Cmax), la concentración en el estado estacionario del producto químico en la sangre (Css), y, lo más importante, el área bajo la curva de concentración-tiempo del fármaco (AUC). AUC expresa una medición válida de la exposición a EE porque mide la cantidad total de EE que alcanza la circulación sanguínea durante un periodo de tiempo (a diferencia de Cmax o Css que únicamente aportan la concentración de estrógenos en la sangre en un instante determinado) [3]. De este modo, AUC ayuda a explicar las diferencias entre los parches y las píldoras en la administración de estrógenos diaria. Así, esta petición se restringe a estudios que comparan los niveles de AUC durante el tiempo (AUC0-t) en la fase estacionaria de los parches y los comprimidos, a menos que se indique lo contrario.
El prospecto original de Ortho-Evra aprobado en 2001 citaba un estudio FC de dosis única que afirmaba que Ortho-Evra aportaba sólo el equivalente de 20mcg de EE al día [4], a pesar de que la compañía ya tenía conocimiento por otro estudio previo a la aprobación de que el parche suministraba más estrógeno que los comprimidos de 30 mcg [5]. En el año 2005, Johnson & Johnson modificó el prospecto de Ortho-Evra para citar un estudio FC posterior a la comercialización del fármaco que halló que la exposición general a EE con los parches, según las mediciones de AUV y Css, era un 55-60% superior que con las píldoras de 35mcg de EE [6].
Lamentablemente, altos niveles de exposición a EE pueden tener consecuencias devastadoras para la salud, particularmente tasas superiores de tromboembolismo venoso (TEV). Un comité asesor de la FDA había llegado a la conclusión en 1988 de que “la eficacia para la prevención del embarazado es la misma para todos los anticonceptivos orales con una dosis igual o superior a 30mcg de estrógeno”, y que los anticonceptivos de altas dosis no “representan un valor único suficiente para la práctica clínica que merezca garantizar su continua disponibilidad como anticonceptivo” [7]. Como consecuencia de esta recomendación, los fabricantes de anticonceptivos orales retiraron voluntariamente del mercado todos los productos de altas dosis (más de 50mcg de estrógeno) [8].
En la actualidad, entre los más de 60 tipos de píldoras en el mercado, sólo dos fármacos genéricos contienen dosis de 50mcg de EE; el resto tienen 35mcg de EE o menos. De hecho, según un libro de texto de farmacología estándar, la anticoncepción hormonal debería consistir en la “dosis mínima de esteroides que proporcionen una cobertura anticonceptiva efectiva. A menudo esto se consigue con una píldora de 30-35mcg de estrógeno, pero los preparados con 20mcg pueden resultar adecuados” [9].
Dado el riesgo innecesario de niveles elevados de estrógenos en la anticoncepción, no es sorprendente que el prospecto de Ortho-Evra tuviera que modificarse de nuevo en 2006 y 2008 para incluir hallazgos procedentes de estudios epidemiológicos que informaban de hasta un doble incremento del riesgo de VTE en mujeres que usaban el parche en comparación con la terapia oral habitual.
Tres estudios documentan que Ortho-Evra aporta una dosis alta de etinil estradiol
PHI-017, el primer estudio del estado estacionario que comparó de forma directa la farmacocinética de Ortho-Evra con la terapia oral, se completó dos años antes de que la FDA aprobase el producto [10]. PHI-017 comparó el parche con un comprimido trifásico (30mcg de EE en los días 1 a 6, 40mcg de EE en los días 7 a 11, y 30mcg de EE en los días 12 a 21/levonorgestrel) e identificó que con los parches había el doble de exposición a EE, lo que equipara los parches a una píldora de EE de 47mcg (ver Apéndice A y B). Finalmente, estos resultados se hicieron públicos como parte de un litigio en curso, estos resultados nunca se han publicado, ni se han mencionado en el prospecto, ni se han puesto en conocimiento del público en general.
Tras la aprobación de la FDA, se efectuaron dos estudios FC comparativos adicionales. El primer estudio, conocido como NED-1, se llevó a cabo en 2003 como requisito post-comercialización de la Agencia Europea de Evaluación de Medicamentos (EMEA) [11,12]. Este estudio mostró una exposición superior al 60% a EE con el parche en comparación con una píldora estándar de EE de 35mcg, y constituyó la base del cambio del prospecto en 2005. El prospecto no mencionó que la exposición a EE (en AUC) con el parche equivalía a una dosis diaria de 56mcg con la píldora (IC 95% =18,9-94,8mcg) (ver Apéndice B). Esto supera la cantidad necesaria para la anticoncepción que el comité asesor de la FDA determinó en 1988, por lo que aumenta el riesgo de efectos adversos sin ningún incremento de la eficacia.
El estudio FC final fue dirigido por van den Heuvel et al., investigadores no afiliados con Johnson & Johnson [13] determinó que había un incremento del 60% del nivel de AUC en Ortho-Evra en comparación con la píldora de EE de 30mcg.
Los lugares de aplicación del parche aportan diferentes cantidades de estrógenos
El único estudio que comparó los niveles de exposición hormonal al administrarse en parche en diferentes partes del cuerpo (abdomen, nalgas, brazo y torso), identificó que el parche en la zona abdominal aportaba aproximadamente un 20% menos de EE que en los otros tres lugares de aplicación [14]. Suponemos que la FDA consideró que esta diferencia resultaba aceptable después de que Johnson & Johnson argumentara que el abdomen aún aportaba una dosis de hormonas “terapéuticamente equivalente” o al menos tan eficaz [15]. Por supuesto, una dosis de igual eficacia de estrógeno es incluso inferior que la dosis del sitio abdominal. Es de destacar que tanto el estudio PHI-017 como el estudio de van den Heuvel et al. sólo midieron el nivel de exposición de estrógeno en la aplicación abdominal. Es posible que los niveles de estrógenos en estos estudios pudieran haber sido incluso superiores si se hubieran incluido los demás lugares de administración.
Ortho-Evra aporta una mayor variabilidad en la exposición a etinil estradiol que las píldoras
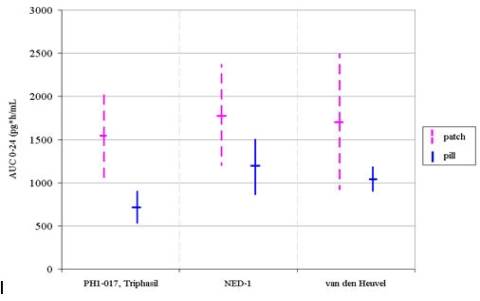
Teóricamente una de las ventajas principales de Ortho-Evra sobre las píldoras anticonceptivas era que Ortho-Evra suministraría una cantidad constante de hormonas mediante la absorción transdérmica sin picos y valles como en la dosis diaria oral con EE. De hecho, las evidencias extraídas de los estudios comparativos de dos anticonceptivos muestran que la variabilidad general de la exposición a estrógenos en los parches es en realidad mayor que la exposición de EE con píldoras (ver Figura 1). Por ejemplo, el intervalo de confianza del 68% (1 desviación estándar) [16] para la exposición de estrógenos del anticonceptivo oral Triphasil en el estudio PHI-017 fue de 534-902 picogramos-hr/ml, mientras que el mismo intervalo para la exposición de estrógenos con Ortho-Evra era de 1.025-2.050 picogramos-hr/ml.
Una comparación de las desviaciones estándar en los tres estudios obtiene como conclusión que, ajustado por el hecho de que las mediciones con mayores medias tienden a tener mayores desviaciones estándar, Ortho-Evra tiene 1,2-3,5 veces más variabilidad en la exposición de estrógenos que las píldoras [17].
Figura 1: Exposición a etinil estradiol en estudios comparativos de dosis múltiples, IC 68% [18] (aplicación del parche únicamente en el abdomen)
La variabilidad en las concentraciones de EE en la fase estacionaria (Css) registrada de los estudios FC de Ortho-Evra también resulta alarmante. Durante el desarrollo de Ortho-Evra, se determinó un rango objetivo de Css para el parche para asegurar la administración de niveles eficaces de hormonas. Este rango objetivo se basó en las concentraciones medias del estado estacionario de estrógenos en mujeres bajo tratamiento con píldoras de 35mcg, o entre 35 y 75 pg/ml [19]. En un estudio controlado de 10 mujeres sanas tratadas con Ortho-Evra, White et al. identificaron niveles medios de Css de 111±63 y 114±59 pg/ml mediante dos tipos diferentes de métodos de ensayo de sangre [20]. El prospecto actual señala que la Css media identificada en los usuarios con parches se encuentra en un rango entre 11,2 y 137 pg/ml [21].
Aumento del riesgo de trombos sanguíneos
Dada la mayor dosis media de EE resultante con Ortho-Evra y la evidencia conocida de que el aumento de la exposición a estrógenos incrementa el riesgo de TEV [22], no resulta sorprendente que los estudios farmacodinámicos (FD) y epidemiológicos sugieran un aumento del riesgo de TEV en mujeres que han recibido tratamiento con el parche en comparación con las píldoras de EE de 20-35mcg.
Estudios farmacodinámicos: Marcadores para el aumento del riesgo de trombos sanguíneos. Los estudios FD miden los cambios químicos que un fármaco provoca en el organismo de un sujeto. Estos estudios son útiles para los marcadores secundarios de seguimiento de potenciales eventos adversos, sobre todo cuando estos eventos son poco comunes o difíciles de estudiar de forma concluyente. El mejor marcador secundario para la evaluación del riesgo de trombosis en usuarios de anticonceptivos hormonales es la globulina de unión a testosterona-estrógeno (Sex Hormone-Binding Globulin o SHGB), porque combina el efecto de los componentes de los estrógenos y la progestina del anticonceptivo para medir el efecto estrogénico total de la píldora, que se relaciona con un aumento del riesgo de trombosi [23,24].
Los tres estudios farmacodinámicos conocidos de Ortho-Evra se efectuaron como ensayos aleatorizados controlados que comparaban, entre otras respuestas bioquímicas, la SHGB de las mujeres que empleaban los parches en comparación con las píldoras. Se extrajeron muestras de sangre al inicio y a la conclusión del último ciclo (es decir, en el estado estacionario) para su análisis. Se empleó una tasa del incremento desde el inicio para comparar las diferencias en los niveles de SHGB.
El primer estudio se llevó a cabo como un componente del estudio NED-1, mencionado anteriormente. La tasa de cambio de SHGB desde el inicio en los sujetos con Ortho-Evra en comparación con los sujetos tratados con EE 35mcg/norgestimato fue del 41% una vez alcanzado el estado estacionario (p=0,0001) [25].
White et al. analizaron los niveles de SHGB en mujeres tratadas con Ortho-Evra en comparación con nueve mujeres del grupo control que tomaron una píldora de EE 35mcg/norgestimato durante tres ciclos. Los sujetos con Ortho-Evra experimentaron un cambio en los niveles de SHGB superior al 73% comparado con el nivel basal (p<0,05) [26]
El estudio publicado más recientemente identificó una diferencia del 33% al comparar los niveles al inicio y a los seis ciclos del uso de anticonceptivos en 32 mujeres tratadas con Ortho-Evra y 33 mujeres que recibieron tratamiento con píldoras de EE de 20mcg/desogestrel (p=0,005) [27]. De este modo, los tres estudios documentaron aumentos estadísticamente significativos del riesgo asociado a SHGB en mujeres tratadas con Ortho-Evra comparado con las tratadas con píldoras anticonceptivas.
Estudios epidemiológicos. En respuesta a las inquietudes surgidas por el aumento de TEV experimentado por mujeres bajo tratamiento con Ortho-Evra, Johnson & Johnson también patrocinó tres estudios de caso-control que comparaban el riesgo de TEV en las que utilizaban Ortho-Evra versus las que utilizaban las píldoras. Los estudios de caso-control tienen como fin detectar la asociación entre un evento adverso poco común (como TEV de etiología idiopática o desconocida en mujeres jóvenes y sanas) y una intervención específica (como el uso de Ortho-Evra). Este método retrospectivo compara los “casos” de mujeres que han experimentado un evento adverso con las mujeres del grupo “control” sin eventos adversos y, en este caso, se determina si el uso de Ortho-Evra es más común entre los casos que entre los controles. Para mitigar las dificultades de los estudios de diseño no aleatorizado, los tres estudios emparejaron casos y controles según características relevantes como la edad y los factores de riesgo de TEV, y analizaron los datos utilizando regresiones logísticas condicionales para controlar otros posibles factores de confusión.
Cole et al. fueron los primeros investigadores que publicaron íntegramente su estudio caso-control sobre Ortho-Evra. Los investigadores emplearon la base de datos de recibos médicos y de farmacia de una compañía aseguradora de salud para identificar los casos de TEV idiopática en mujeres a las que se les había recetado recientemente Ortho-Evra o píldoras de EE de 35mcg/norgestimato. Los casos de TEV se confirmaron con los resúmenes del historial médico. A continuación se seleccionaron al azar cuatro controles sin TEV de la misma base de datos y emparejaron cada caso por año de nacimiento, factores de riesgo de TEV y los antecedentes particulares de exposición al anticonceptivo relevante. Este estudio reveló que, en comparación con las mujeres sin TEV idiopática, las mujeres con TEV idiopática tuvieron una probabilidad 2,4 superior de haber recibido tratamiento con Ortho-Evra que con la píldora de EE/norgestimato de 35mcg (IC 95%=1,1-5,5).[28]
El segundo estudio, por Jick et al., empleó una metodología ligeramente diferente y no identificó un incremento de la asociación entre Ortho-Evra y TEV idiopático no fatal en comparación con la píldora de EE de 35mcg/norgestimato (Odss ratio=1,0; IC 95%=0,7-1,5) [29]. Este estudio extrajo su población de una base de datos de una compañía aseguradora diferente, y a diferencia del estudio Cole, sólo incluyó mujeres que recibieron tratamiento por primera vez con uno de los fármacos del estudio, excluyeron los casos de TEV fatal y no confirmaron los casos de TEV con los registros médicos. La FDA utilizó estos dos estudios de caso-control como base para la revisión del prospecto del 2006.
Parte de la información procedente de un tercer estudio de caso-control, aún sin publicar, se hizo pública para incluirla en la revisión del prospecto de Ortho-Evra en 2008 [30]. Este estudió también fue dirigido por el grupo de Jick y empleó una metodología similar al estudio previo. En esta ocasión los investigadores compararon Ortho-Evra con la píldora de EE de 30mcg/levonorgestrel e identificaron un aumento del riesgo, pero no lo suficiente para que fuese estadísticamente significativo, de TEV idiopático no fatal para los sujetos tratados con Ortho-Evra (OR=2,0; IC 95%= 0,9-4,1).
El prospecto de 2008 concluyó que, “se desconoce si hay cambios en el riesgo de eventos adversos graves a partir de las diferencias en los perfiles farmacocinéticos de EE en mujeres tratadas con ORTHO EVRA® en comparación con mujeres tratadas con anticonceptivos orales con 35mcg de EE”. Aunque estos estudios epidemiológicos no son concluyentes por sí solos, unidos a los datos FC y FD, sugieren un riesgo de TEV mayor que el admitido en el prospecto.
Más efectos adversos y mayor tasa de interrupción
Además de los casos de TEV, en dos ensayos clínicos de gran tamaño sobre la eficacia de Ortho-Evra en comparación con las píldoras se ha observado un aumento de las tasas de otros efectos adversos. Audet et al. aleatorizaron 1.495 mujeres para la recibir los parches o una píldora trifásica (EE 30mcg en los días 1 a 6, EE 40mcg en los días 7 a 11, y EE 30mcg en los días 12 a 21/levonorgestrel). Un tercio de los participantes se inscribieron para la recepción de 13 ciclos y el resto se inscribió en seis ciclos [31]. Urdl et al. aleatorizaron de forma similar a 1.517 mujeres para recibir el parche o una píldora monofásica (EE 20mcg/desogestrel durante 21 días) durante 6 ó 13 ciclos de tratamiento [32]. Ambos ensayos hallaron que la probabilidad de efectos secundarios dolorosos era estadísticamente más significativa en sujetos tratados con Ortho-Evra que con píldoras anticonceptivas. Por ejemplo, Audet y Urdl identificaron que entre el 19% y 25% de las mujeres tratadas con Ortho-Evra en ambos ensayos experimentaron molestias en las mamas, tres veces más que los sujetos tratados con píldoras (OR=3,09; IC 95%=2,26-4,22], OR=2,98; IC 95%=2,29-3,90], respectivamente). Audet et al. identificaron que el 13% de las tratadas con Ortho-Evra experimentaron dolores menstruales severos (OR=1,43; IC 95%=1,03-1,99). Urdl et al. también identificaron un aumento de casos de náuseas (OR=2,08; IC 95%=1,46,2,95) y vómitos (OR=1,88; IC 95%=1,12-3,16) (Ver Apéndice C. En los casos donde no se notificaron los efectos secundarios, no se obtuvieron resultados o bien estos no eran significativos).
Los efectos secundarios influyeron para que las mujeres interrumpieran el tratamiento con un producto particular. La probabilidad de que las mujeres aleatorizadas a recibir el parche abandonaran los ensayos de eficacia anteriormente mencionados fue consistentemente mayor que en aquellas asignadas al tratamiento con píldoras. Por ejemplo, Audet et al. hallaron que el 12% de todas las mujeres aleatorizadas a Ortho-Evra abandonaron el estudio debido a los eventos adversos, en comparación con el 5% de mujeres que tomaron la píldora trifásica (OR=2,27; IC 95%=1,59-3,25). Urdl et al. hallaron tasas similares de interrupción debido a los eventos adversos (OR=2,01; IC 95%=1,38-2,95). En estos estudios, la interrupción del tratamiento con Ortho-Evra por cualquier motivo (incluidos los efectos adversos) fue de 1,45 a 1,58 (p<0,05) veces más probable en comparación con la píldora. Otros estudios no aleatorizados o controlados efectuados para observar el uso típico de los anticonceptivos en poblaciones de alto riesgo sugieren una menor aceptabilidad por parte del usuario y una tasa mayor de embarazos con el parche en comparación con la píldora [33,34]. Los estudios sobre las píldoras han demostrado que cualquier interrupción del tratamiento conduce a un periodo sin uso de anticonceptivos y un aumento consecuente del riesgo de embarazo [35].
Misma eficacia que píldoras a pesar de la mayor adherencia
Audet et al. y Urdl et al. también midieron la adherencia al tratamiento, definido para las píldoras como la proporción de ciclos completados con 21 días consecutivos de ingesta del fármaco y para el parche como que no se llevase el mismo parche durante más de 7 u 8 días, respectivamente. Los resultados de la adherencia no incluyeron los ciclos potenciales de mujeres que decidieron abandonar el estudio. Ambos estudios hallaron que la adherencia al tratamiento fue mayor en los sujetos tratados con parches que en los sujetos tratados con píldoras (OR=2,05; IC 95%=1,83-2,29] y OR=2,76; IC 95%=2,35-3,24]. Sin embargo, esta ventaja no se tradujo en un descenso estadísticamente significativo de embarazos (OR=0,57; IC 95%=0,18-1,77] y OR=1,49; IC 95%=0,30-7,53) en estudios de muestras insuficientes para poder detectar diferencias estadísticamente significativas. Aunque el aumento de la adherencia de las mujeres tratadas con Ortho-Evra en comparación con las píldoras constituye uno de los reclamos de Johnson & Johnson, los resultados similares de embarazos entre las usuarias que continuaron en el ensayo ponen en duda la relevancia del resultado.
Fundamento para la retirada paulatina del mercado
Sin una eficacia adicional y con un considerable riesgo extra que supera los niveles aceptados para las píldoras anticonceptivas, Ortho-Evra constituye una mala elección para las mujeres. En cualquier caso, el uso de los parches es claramente más eficaz que no usar un método de anticoncepción. La retirada de cualquier anticonceptivo del mercado conlleva el riesgo de que alguna proporción de los usuarios no sustituya de forma inmediata su anticoncepción con un método que sea al menos tan efectivo como el producto prohibido. Lo ideal sería que Johnson & Johnson comercializara inmediatamente una reformulación más segura del parche que suministrara niveles inferiores y menos variables de estrógenos, aunque esto no ha sucedido en los siete años de comercialización de Ortho-Evra. Por lo tanto, Public Citizen solicita un periodo de transición de seis meses en el que Ortho-Evra estará disponible comercialmente para continuar el tratamiento hasta que las mujeres puedan concertar una cita con su facultativo y buscar un método anticonceptivo alternativo.
Conclusión
Los beneficios teóricos de Ortho-Evra sobre otras opciones anticonceptivas incluían una menor variabilidad en la administración de estrógenos y una mejor adherencia al tratamiento. Por desgracia, Ortho-Evra supone algo más que riesgos teóricos. Los sujetos que reciben tratamiento con Ortho-Evra están expuestos a niveles de estrógenos que, como media, podrían compararse a una píldora de EE de 56mcg, pero podría oscilar ampliamente entre las mujeres expuestas, de 18mcg a 95mcg de EE. Los eventos adversos documentados incluyen un incremento del 200% de casos de tromboembolismo venoso y una mayor variedad de efectos secundarios dolorosos. Por último, a pesar de la mejor adherencia, no existen diferencias en el número embarazos en comparación con las píldoras. Si Ortho-Evra hubiera sido diseñado como una píldora, es poco probable que hubiera sido aprobada debido a su mayor contenido de estrógenos. La lógica preocupación sobre seguridad relativa de la exposición a dosis variables y elevadas de estrógenos inclina la balanza de riesgos y beneficios contra la disponibilidad de Ortho-Evra como anticonceptivo.
Impacto medioambiental
La retirada de Ortho-Evra del mercado será beneficioso para el medio ambiente, ya que los parches usados aún contienen considerables cantidades de estrógenos (80% de la dosis original de estrógeno) que representa un riesgo mayor de fuga y contaminación que los comprimidos [36].
Atentamente
Eunice Yu, Investigadora
Sidney M. Wolfe, Director Ejecutivo
Grupo de Investigación en Salud de Public Citizen
Apéndice A: Resumen de estudios farmacocinéticos de Ortho-Evra de dosis múltiples
Estudios precomercialización
PHI-005 [37]
Este estudio abierto de Ortho-Evra se llevó a cabo en 12 mujeres para determinar el perfil FC de dos parches consecutivos. El primer parche se aplicó durante 7 días y el segundo parche se aplicó durante 10 días para evaluar el efecto FC de portar de forma inadecuada el parche durante 3 días más que el intervalo de dosis prescrito de 7 días. Todos los parches se colocaron en el abdomen y se extrajeron muestras de sangre a lo largo del estudio. Se registraron los resultados AUC en el 7º día del segundo parche, lo que proporciona mediciones de AUC de dosis múltiples que son comparables a otros estudios (ver Tabla A1)
PHI-013 [38]
El estudio PHI-013 realizó seguimiento a 24 mujeres durante tres ciclos de aplicación del parche y constituye el estudio FC de mayor duración realizado sobre Ortho-Evra. Las mujeres fueron aleatorizadas para portar el parche de acuerdo con las instrucciones del producto en las nalgas o el abdomen durante los periodos de medición (semana 1 del ciclo 1 y semanas 1,2 y 3 del ciclo 3), pero eran libres de portar el parche en cualquiera de las cuatro zonas anatómicas durante las semanas sin medición (ver Tabla A1).
Tabla A1: Exposición de etinil estradiol procedente de estudios FC de
dosis múltiples de Ortho-Evra
|
Estudio |
Lugar de aplicación |
Duración de la aplicación del parche |
Tamaño muestral |
AUC0-168h media (pg*h/ml) |
DE |
|
PHI-005 |
Abdomen |
7+10 días |
12 |
8353 |
3098 |
|
PHI-013 |
Abdomen |
3 x 21 días |
12 |
12139 |
3241 |
|
PHI-013 |
Nalgas |
3 x 21 días |
12 |
8840 |
5176 |
|
PHI-017* |
Abdomen |
21 días |
12 |
10761 |
3589 |
|
NED-1 |
Abdomen y nalgas |
2 x 21 días |
31 |
12974 |
4295 |
|
Van den Heuvel et al. |
Abdomen |
21 días |
8 |
11933 |
1833 |
*Triphasil sólo en brazo
PHI-017 [39]
El estudio PHI-017 comparó el parche Ortho-Evra en abdomen con tres anticonceptivos orales: Triphasil (un régimen trifásico de levonorgestrel y 30mcg de EE durante seis días, 40mcg de EE durante cinco días, y 30mcg de EE durante 10 días), Alesse (levonorgestrel y 20mcg de EE), y Mercilon (desogestrel y 20mcg de EE). Treinta y seis mujeres fueron aleatorizadas para recibir una de las tres píldoras en un estudio cruzado efectuado en mujeres que recibieron tratamiento por primera vez, o bien con la píldora asignada o con el parche, seguido por un periodo sin tratamiento durante un mínimo de 28 días y un tratamiento alternativo. El grupo Evra-Triphasil recibió el ciclo completo de 21 días de píldoras y Ortho-Evra durante 21 días, mientras que los grupos Evra-Alesse y Evra-Mercilon recibieron tratamiento solamente siete días (una píldora al día o un parche semanal) de cada tratamiento. Sólo se extrajeron muestras de sangre durante la última semana del grupo Evra-Triphasil y durante la semana para los grupos Evra-Alesse y Evra-Mercilon (Figura 1).
Los resultados mostraron que las mujeres tenían niveles de AUC en el estado estacionario de EE que eran el doble de altos con Ortho-Evra en comparación 30-40mcg de Triphasil. La exposición a EE fue tres veces superior en las tratadas con Ortho-Evra en comparación con 20mcg de EE de Alesse y Mercilon. La conclusión del estudio sugirió que estas comparaciones no eran válidas debido a que la exposición a EE procedente de las píldoras en este estudio era inusualmente baja en comparación con sus valores de referencia. Al mismo tiempo, los resultados de Ortho-Evra de este estudio son similares a estudios previos. Así, “la exposición por debajo de los esperado de etinil estradiol tras la administración del anticonceptivo oral condujo a una mayor exposición relativa a etinil estradiol tras la dosificación con EVRA”. Según este razonamiento, las conclusiones enviadas a la FDA dicen, “los datos procedentes de este y otros estudios indican que EVRA es comparable en exposición y niveles alcanzados de estrógenos a anticonceptivos orales de 35mcg”.
Esta afirmación intenta realizar una comparación falsa entre los resultados de AUC del parche en este estudio con estudios anteriores de píldoras llevados a cabo en diferentes poblaciones, mientras que ignora los resultados de un grupo comparador aleatorizado concurrente que se incluyó en el diseño del estudio. De hecho, el diseño del estudio cruzado exige que la compañía demuestre por qué las mismas mujeres obtienen resultados sustancialmente diferentes con los parches en comparación con las píldoras. Aunque este estudio sugirió un nivel más alto de exposición de EE con los parches en comparación con las píldoras antes de su aprobación (el estudio se completó en junio de 1999, casi dos años antes de su aprobación por la FDA), el prospecto aprobado por la FDA para Ortho-Evra nunca ha mencionado el estudio PH1-017 o sus resultados. Además, la FDA no menciona este estudio en sus revisiones farmacológicas clínicas ni ha sido reconocido por Johnson & Johnson en sus últimos comunicaciones con la FDA. No resulta sorprendente que tampoco se haya publicado.
Estudios post-comercialización
NED-1 [40,41]
En el primer estudio FC post-comercialización, 36 mujeres fueron aleatorizadas en un estudio abierto y cruzado que comparó Ortho-Evra con Cilest (norgestimato y 35mcg de EE). Las mujeres recibieron tratamiento con los parches o las píldoras durante dos ciclos seguidos con un periodo de reposo farmacológico de 28 días y luego recibieron el tratamiento alternativo durante dos ciclos. Cada ciclo constó de 21 días consecutivos de tratamiento seguidos de siete días sin tratamiento. Asimismo se aleatorizó la colocación del parche en las nalgas o el abdomen. Se extrajeron las muestras de sangre durante la semana uno del ciclo uno y en la semana tres del ciclo dos.
Como se mencionó anteriormente, este estudio demostró que la exposición media de EE en la fase estacionaria del parche fue un 60% superior que la AUC media de las píldoras de 35mcg de EE, la dosis anticonceptiva típica más alta en el mercado (ver Figura 1).
Van den Heuvel et al. [42]
En este estudio post-comercialización independiente, 24 mujeres tomaron en primer lugar una píldora, Microgynon (levonorgestrel y 30mcg de EE) durante dos a ocho semanas antes de un periodo de siete días sin tratamiento y se aleatorizaron para continuar con 21 días de tratamiento con el parche NuvaRing o bien Microgynon. Todas las mujeres tratadas con el parche lo portaron en el abdomen. Se extrajeron muestras de sangre a lo largo de todo el tratamiento (Figura 1).
Apéndice B: Relación lineal AUC-dosis
La afirmación de que Ortho-Evra contiene 56mcg de etinil estradiol procede del estudio NED-1, en el cual los niveles de sangre de EE en mujeres tratadas con el parche se compararon con sus niveles de EE cuando fueron tratadas con píldoras de 35mcg. El estudio concluyó que Ortho.-Evra exponía a las mujeres a un 60% más de EE, así un 60% más de 35mcg equivalen a una dosis de estrógenos de 56mcg. Los expertos de Johnson & Johnson y sus propios estudios han confirmado la relación lineal entre la AUC de estrógenos y la dosis de la píldora equivalente [43]. El estudio PHI-017 explica que “la dosis estimada de EE (en comparación con OC) para Evra en un periodo de 24 horas puede calcularse mediante una ecuación de regresión” [44], derivada del trazado de las AUC de varias dosis de píldoras (Ver Figura 1B). El estudio NED-1 utiliza de nuevo esta ecuación de regresión lineal para calcular la dosis de EE equivalente para todos los estudios FC realizados sobre Ortho-Evra [45]. La tabla 1B muestra que la dosis equivalente de EE suministrada de los lotes comerciales de parches Ortho-Evra en el estado estacionario tienen una media de 56mcg de EE (IC 95%: 18,9-94,8) mediante esta ecuación de regresión lineal.
Figura 1B: Dosis de EE vs. AUC0-24 para seis píldoras anticonceptivas orales [46]
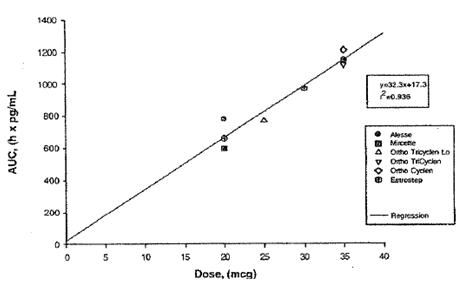
No está claro por qué el cambio del prospecto de Ortho-Evra en el que se indicaba el nivel verdadero de exposición a un ingrediente activo en un producto se produjo en el año 2005, casi cuatro años después de su aprobación inicial. Es posible que los lotes más pequeños de parches que se desarrollaron inicialmente para los ensayos clínicos aportaran en realidad menos EE de media que aquellos que se fabricaron con posterioridad en una mayor escala para su comercialización. Una comparación de los resultados de AUC procedentes de estudios de dosis múltiples de Ortho-Evra muestran que los parches comerciales exponían a las mujeres a una media de 12mcg más de EE al día que los parches de los ensayos clínicos (no significativo, p>0,05). Existen evidencias limitadas disponibles al público de que Johnson & Johnson ha tenido otros problemas con la fabricación de Ortho-Evra. Por ejemplo, las versiones europeas y canadienses de los parches sólo contienen 0,6mg de EE, sin embargo, afirman su bioequivalencia con el parche de 0,75mg comercializado en EE.UU. [47,48].
Tabla 1B: Dosis de etinil estradiol equivalente procedente de estudios FC de dosis múltiples de Ortho-Evra
|
Estudio [49] |
Lote de fabricación |
Lugar de aplicación |
Dosis de EE [50] (mcg/día) |
IC 95% |
Dosis de EE media [51] (mcg/día) |
|
|
Inferior |
Superior |
|||||
|
PHI-005 |
Clínica Fase 2 |
Abdomen |
36,4 |
9,0 |
63,8 |
|
|
PHI-013 |
Clínica Fase 3 |
Abdomen |
53,2 |
24,5 |
81,8 |
|
|
PHI-013 |
Clínica Fase 3 |
Nalgas |
38,6 |
0,0 |
84,3 |
|
|
PHI-017* |
? |
Abdomen |
47,1 |
15,3 |
78,8 |
43,8 |
|
NED-1 |
Comercial |
Abdomen y nalgas |
56,8 |
18,9 |
94,8 |
|
|
Van den Heuvel et al. |
Comercial |
Abdomen |
52,2 |
36,0 |
68,5 |
55,9 |
*Triphasil sólo en brazo
Apéndice C: Tasas de interrupción y eventos adversos con el parche en comparación con un anticonceptivo oral [52]
|
|
Estudio |
Audet 2001 |
Urdl 2005 |
|
Píldora de referencia |
EE 30/40/30mcg / LNG 50/75/125mcg |
EE 20mcg / DSG 150mcg |
|
|
Tamaño muestral (Ortho Evra/ Píldora) |
856 / 639 |
846 / 643 |
|
|
Interrupción – General |
% O-E |
30% |
20% |
|
% Píldora |
21% |
14% |
|
|
Odds Ratio |
1,58 [1,25, 1,99] |
1,45 [1,11, 1,90] |
|
|
Interrupción – Debido a eventos adversos |
% O-E |
12% |
10% |
|
% Píldora |
5% |
5% |
|
|
Odds Ratio |
2,27 [1,59, 3,25] |
2,01 [1,38, 2,95] |
|
|
Molestias en mamas |
% O-E |
19% |
25% |
|
% Píldora |
6% |
9% |
|
|
Odds Ratio |
3,09 [2,26, 4,22] |
2,98 [2,29, 3,90] |
|
|
Dolor menstrual severo |
% O-E |
13% |
5% |
|
% Píldora |
10% |
5% |
|
|
Odds Ratio |
1,43 [1,03, 1,99] |
1,15 [0,72, 1,83] |
|
|
Náuseas |
% O-E |
20% |
12% |
|
% Píldora |
18% |
6% |
|
|
Odds Ratio |
1,14 [0,88, 1,49] |
2,08 [1,46, 2,95] |
|
|
Vómitos |
% O-E |
No notificado |
5% |
|
% Píldora |
3% |
||
|
Odds Ratio |
1,88 [1,12, 3,16] |
Referencias:
1. Drug Topics “Top 200 brand drugs by retail dollars in 2007” Accessed March 20, 2008. drugtopics.modernmedicine.com/drugtopics/article/articleList.jsp?sort=null&pageNo=1&start=0&categoryId=7604&article
Type=%20&numberOfDays=%20&contentType=
2. Johnson & Johnson Pharmaceutical Research Institute. ”Clinical Study Report Protocol NRGEEP-PHI-014; Phase 1.” May 4, 1999. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.583 and Johnson & Johnson Pharmaceutical Research Institute. “Clinical Study Report Protocol NRGEEP-PHI-017; Phase 1” June 11, 1999. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.591.
3. Shargel L and Yu A. Applied Biopharmaceutics and Pharmacokinetics. 1985, Appleton-Century-Crofts/Prentice-Hall. East Norwalk, Connecticut. P.132.
4. Ortho-Evra label. November 20, 2001. Available at www.fda.gov/cder/foi/label/2001/21180lbl.pdf.
5. Johnson & Johnson Pharmaceutical Research Institute. ”Clinical Study Report Protocol NRGEEP-PHI-017; Phase 1.” June 11, 1999. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.591.
6. Ortho-Evra label. November 10, 2005. Available at www.fda.gov/cder/foi/label/2005/021180s019lbl.pdf.
7. Food and Drug Administration, Transcript of the Fertility and Maternal Health Drugs Advisory Committee. Volume II. January 15, 1988, P.166.
8. Ortho Pharmaceutical Corporation., “Dear Doctor letter.” March 30, 1988. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No., 1:06-40000, Deposition Ex. 674.
9. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 10th Ed. 2001. McGraw Hill.
10. Johnson & Johnson Pharmaceutical Research Institute. ”Clinical Study Report Protocol NRGEEP-PHI-017; Phase 1.” June 11, 1999. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.591.
11. Johnson & Johnson Pharmaceutical Research and Development LLC. ”Clinical Study Report Protocol PRI/EDN-NED-1; Phase 1.” March 10, 2003. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.629.
12. Devineni D, Skee D, Vaccaro N, Massarella J, Janssens L, LaGuardia KD, Leung AT. J Clin Pharmacol 2007; 47:497-509.
13. van den Heuvel MW, van Bragt AJM, Alnabawy AKM, Kaptein MCJ. Contraception 2005;72:168-174.
14. Abrams LS, Skee DM, Natarajan J, Wong FA, Anderson GD. Br J Clin Pharmacol 2002; 53:141-146.
15. Abrams LS, Skee DM, Natarajan J, Wong FA, Anderson GD. Br J Clin Pharmacol 2002; 53:141-146.
16. A 68% confidence interval means there is a 68% chance of the true parameter (in this case the mean) falling within the interval. A wider interval gives greater confidence but in this case is used to indicate the large range of AUC levels in Ortho-Evra compared to pill users.
17. Calculated as a ratio of the percent coefficient of variation (%CV) of Ortho-Evra over the pill. %CV is the standard deviation divided by the mean, expressed as a percentage.
18. Dotted lines show variation in AUC data from the patch worn on the abdomen only (other sites are approximately equivalent, or higher). Solid lines show variation in AUC from the same study of the pill. Sample sizes in each group are approximately equal.
19. Ortho-Evra label. November 20, 2001. Available at www.fda.gov/cder/foi/label/2001/21180lbl.pdf.
20. White T, Ozel B, Jain JK, Stanczyk FZ. Contraception 2006;74:293-296.
21. Ortho-Evra label. January 18, 2008. Available at www.fda.gov/cder/foi/label/2008/021180s026lbl.pdf
22. van Vliet HAAM, Frolich M, Christella M, Thomassen LGD, Doggen CJM, Rosendaal FR, Rosing J, Helmerhorst FM. Hum Reprod 2005;20:563-568.
23. van Vliet HAAM, Frolich M, Christella M, Thomassen LGD, Doggen CJM, Rosendaal FR, Rosing J, Helmerhorst FM. Hum Reprod 2005;20:563-568.
24. Van Rooijen M, Silveira A, Hamsten A, Bremme K. Am J Obstet Gynecol 2004;190:332-337.
25. From study NED-1, p-values not published in Devenini et al. (2007)
26. White T, Ozel B, Jain JK, Stanczyk FZ. Contraception 2006;74:293-296.
27. Kluft C, Meijer P, LaGuardia KD, Fisher AC. Contraception 2008;77:77-83.
28. Cole JA, Norman H, Doherty M, Walker AM. Obstet Gynecol. 2007; 109: 339-346.
29. Jick S, Kaye J, Li L, Jick H. Contraception 2007; 76:4-7.
30. Johnson & Johnson Pharmaceutical Research & Development, L.L.C. ClinicalTrials.gov ID: NCT00511784. Accessed on March 11, 2008 clinicaltrials.gov/ct2/show/NCT00511784?term=ortho+evra&rank=10
31. Audet M, Moreau M, Koltun WD, Waldbaum AS, Shangold G, Fisher A, Creasy GW. JAMA 2001;285:2347-2354
32. Urdl W, Apter D, Alperstein, Koll P, Schonian S, Bringer J, Fisher A, Preik M. Obstet Gynecol 2005;121: 202-210.
33. Thurman AR, Hammond N, Brown HE, Roddy ME. J Pediatr Adolesc Gynecol 2007;20:61-65.
34. Bakhru A, Stanwood N. Obstet Gynecol 2006;108:378-86.
35. Rosenberg MJ, Waugh MS, Long S. J Reprod Med 1995; 40:355-60.
36. Prescrire Editorial Staff. “Do Evra contraceptive patches represent an advance?” Prescrire Int 2004;13:123-135.
37. Abrams LS, Skee DM, Wong FA, Anderson NJ, Leese PT. J Clin Pharmacol 2001; 41:1232-1237.
38. Abrams LS, Skee DM, Natarajan J, Wong FA, Lasseter KC. Contraception 2001;64:287-294.
39. Johnson & Johnson Pharmaceutical Research Institute. “Clinical Study Report Protocol NRGEEP-PHI-017; Phase 1” June 11, 1999. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.591.
40. Johnson & Johnson Pharmaceutical Research and Development LLC. “Clinical Study Report Protocol PRI/EDN-NED-1; Phase 1” March 10, 2003. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.629.
41. Devineni D, Skee D, Vaccaro N, Massarella J, Janssens L, LaGuardia KD, Leung AT. J Clin Pharmacol 2007;47:497-509.
42. van den Heuvel MW, van Bragt AJM, Alnabawy AKM, Kaptein MCJ. Contraception 2005; 72:168-174.
43. Deposition of Frank Z. Stanczyk PhD. in re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex. H.
44. Johnson & Johnson Pharmaceutical Research Institute. “Clinical Study Report Protocol NRGEEP-PHI-017; Phase 1” June 11, 1999. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.591. pp 32.
45. See study NED-1 Table 24. But this table uses only single-dose data, even when multiple-dose data are available
46. Johnson & Johnson Pharmaceutical Research Institute. “Clinical Study Report Protocol NRGEEP-PHI-017; Phase 1” June 11, 1999. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.591.
47. Prescribe Editorial Staff “Evra dose change: an air of secrecy” Prescribe Int 2007;16(89):107.
48. Health Canada “New Safety Information about Evra Transdermal System” November 21, 2006. Accessed on March 31, 2008 www.hc-sc.gc.ca/dhp-mps/medeff/advisories-avis/prof/2006/evra_hpc-cps_e.html
49. Data on manufacturing lots comes from study NED-1 Table 24 pp.87. Data used to calculated EE dose and CI taken from the following published and unpublished sources.
PHI-005: Abrams LS, Skee DM, Wong FA, Anderson NJ, Leese PT. J Clin Pharmacol 2001; 41:1232-1237.
PHI-013: Abrams LS, Skee DM, Natarajan J, Wong FA, Lasseter KC. Contraception 2001;64:287-294.
PHI-017: Johnson & Johnson Pharmaceutical Research Institute. “Clinical Study Report Protocol NRGEEP-PHI-017; Phase 1” June 11, 1999. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.591.
NED-1: Johnson & Johnson Pharmaceutical Research and Development LLC. ”Clinical Study Report Protocol PRI/EDN-NED-1; Phase 1” March 10, 2003. In re: Ortho Evra Products Liability Litigation, MDL 1742, N.D. Ohio, Case No. 1:06-cv-40000, Ex.629.
50. Equivalent EE OC Dose = (AUC24h-17.3)/32.3; equation from PHI-017 and NED-1 submissions to the FDA
51. La media es ponderada según el tamaño de la muestra de los estudios.
52. Data from: Audet M, Moreau M, Koltun WD, Waldbaum AS, Shangold G, Fisher A, Creasy GW. JAMA 2001;285:2347-2354 and Urdl W, Apter D, Alperstein, Koll P, Schonian S, Bringer J, Fisher A, Preik M. Obstet Gynecol 2005;121:202-210. Odds ratio data from: Lopez LM, Grimes DA, Gallo MF, Schulz KF. Cochrane Database Syst Rev 2008;1:CD003552.