Entrevista Ensayos clínicos en América Latina: Riesgos y dilemas éticos de la “Innovación científica”
Salud y Fármacos
En América Latina, la expansión de los ensayos clínicos patrocinados por la industria farmacéutica transnacional ha planteado serios interrogantes éticos y regulatorios.
Colombia se perfila como centro regional para esta investigación, en este panorama complejo y lleno de tensiones entre innovación, economía y bioética, proponemos una serie de reflexiones, que las presentamos a modo de entrevista. Natalia Castrillón entrevista a Nuria Homedes (NH) y a Bernardo Useche (BU).
- ¿Por qué hay que hacer tantos ensayos clínicos, es tan significativo el aporte de la “Innovación científica”?
NH. Se desconoce el número de ensayos clínicos que a nivel global se inician en un año determinado, pero son miles. Solo en Colombia cada año se registran más de 60 ensayos clínicos financiados por la industria farmacéutica. También sabemos que las agencias reguladoras de referencia (FDA, EMA), aprueban – en un buen año- 40 o 50 productos nuevos, pero de esos – según las agencias reguladoras y los boletines independientes de medicamentos- solo uno o dos añaden valor terapéutico a los tratamientos existentes.
Hay años en que ninguno de los medicamentos recién aprobados es superior al tratamiento estándar, y hay años en que se produce un medicamento verdaderamente revolucionario. Lo que sí suele suceder es que estos medicamentos nuevos son más caros que los existentes.
Otra cosa a tener en cuenta es que, según datos de la industria, solo alrededor del 10% de los productos que se testan en ensayos clínicos en humanos (Fase I a Fase III) acaban siendo aprobados por las agencias reguladoras de referencia. Es decir, muchas moléculas se quedan por el camino. Estos porcentajes varían por especialidad (son más positivos para enfermedades respiratorias, pero peores en el caso de productos oncológicos). Estas cifras podrían mejorar con el uso de farmacogenómica.
Ahora bien, la mayoría de los ensayos clínicos que se hacen en AL son de Fase III, y el porcentaje de éxito de estos ensayos es de alrededor del 50%, por lo que podemos asumir que solo la mitad de los productos experimentales que se testan en ensayos de Fase III en América Latina serán aprobados por las agencias reguladoras de referencia – y si se tiene en cuenta que solo la mitad de los participantes en los ensayos clínicos reciben el medicamento experimental (la otra mitad suele recibir el tratamiento habitual o un placebo) pues queda claro que solo una parte de los participantes en ensayos clínicos se benefician.
También hay que tener en cuenta que algunos efectos adversos de los medicamentos no se conocen hasta que aumenta el número de personas que los utilizan, por eso hay quienes recomiendan que no se consuman medicamentos nuevos hasta 7 años después de su aprobación, para dar tiempo a que se conozca su perfil de seguridad [1]. La información disponible indica que la mayoría de los productos que resultan ser inseguros cuando los utiliza la población general, se retiran del mercado durante ese periodo de tiempo.
- ¿Cuáles son los riesgos para los participantes?
NH, El riesgo más importante para los participantes es que se está utilizando un producto experimental. Es decir, los participantes son parte de un experimento. Esta palabra ha desaparecido del léxico que se utiliza al hablar de ensayos clínicos, y no solo en los países hispanoparlantes [2], por las connotaciones negativas que puede tener el decir que se está experimentando en humanos. Solemos referirnos a los ensayos clínicos como protocolos, proyectos, estudios…
El otro riesgo que identificó el personal de la agencia reguladora de ensayos clínicos de Perú es que los participantes en los ensayos clínicos no siempre se adhieren a las indicaciones del investigador principal o del coordinador del estudio. No lo hacen con mala intención, al contrario, lo hacen para no molestar y por ignorancia. Desconocen la importancia de adherirse a las instrucciones y cómo esto afecta los resultados… y a veces, si se sienten mal después de tomar el medicamento por la noche o los fines de semana, para no molestar al equipo de investigación no dicen nada [3].
Lo que la agencia reguladora de Perú descubrió es que muchos de los participantes en los ensayos clínicos consumían otros medicamentos o productos de medicina tradicional y no se lo decían al médico investigador. A veces se iban a las salas de emergencia de hospitales y no les decían que estaban participando en un ensayo clínico, otras veces el participante tomaba la mitad de la dosis… Esta información no siempre la compartían con el investigador, porque no querían que los echaran del “proyecto” (protocolo) [4].
Hay que decir que los participantes en ensayos clínicos suelen estar muy satisfechos con el trato que reciben, con la privacidad con la que se les trata, y con las pruebas que les hacen… Se sienten bien cuidados… y no son conscientes de los riesgos que conlleva el producto experimental o las pruebas clínicas que se les realizan.
- ¿No dice el consentimiento informado que deben acudir al centro de investigación cuando estas situaciones se presentan?
NH. Si, el consentimiento informado lo dice, pero muchos participantes en ensayos clínicos firman el consentimiento informado sin haberlo leído, y si lo han leído es reconocido que no lo entienden, hacen lo que se les dice porque se fían de su médico…y otras veces hay una inducción indebida por parte del reclutador. En el estudio de Perú algunos entrevistados dijeron: ¨el médico me dijo que si fuera su hija me inscribiría en el ensayo… que si participaba en el ensayo me curaría, … que si participaba en el ensayo tendría acceso al mejor tratamiento, si no, solo al del sector público¨ [5].
Los participantes a veces leen el consentimiento cuando llegan a su casa, pero los consentimientos son cada vez más largos, incluyen términos que los participantes no entienden, incluso miembros de CEI dicen que no los entienden. Es muy difícil transmitir a los participantes todo lo que deben de saber en un solo documento.
La mayoría de los entrevistados en Perú sabían que si se sentían mal tenían que ir al centro de investigación, pero no lo hicieron por no molestar o porque no asociaron su problema con la participación en el ensayo.
- ¿Quién vigila el comportamiento del paciente que está participando en los ensayos clínicos?
NH. La regulación de la mayoría de los países de América Latina exige que los CEI den seguimiento a los participantes en ensayos clínicos, pero eso se hace en base a los documentos que les entrega el investigador principal, el patrocinador o las agencias reguladoras. No hablan con los participantes, por lo que no tienen acceso a información importante como la que descubrió la agencia reguladora de Perú.
Solo dos hospitales de una provincia de Buenos Aires han contratado a trabajadoras que verifican que los participantes entiendan el consentimiento informado antes de firmarlo, pero después no dan seguimiento al paciente. Este seguimiento del paciente es muy importante para asegurar la integridad de la información que se recaba durante los ensayos clínicos, y también para proteger a los participantes. Hay CEI que tienen interés en este tipo de actividad, pero no tienen tiempo para hacerlo. Además es una actividad que no está remunerada.
- ¿Cómo se aseguran los médicos de que los pacientes se adhieren a sus recomendaciones y frecuencias de dosificación?
NH. Esto depende de lo que diga el protocolo. Hay muchas metodologías para tratar de documentar la adherencia al tratamiento. El asunto es que el participante quiere complacer al investigador porque en general los participantes quieren permanecer en el estudio… eso hace que el paciente no siempre comparta toda la información con el investigador… Los que fueron entrevistados en el estudio de Perú contaron cosas, pero dijeron a los entrevistadores, “por favor no se lo diga al investigador porque me echará del estudio”.
La persona que entreviste a los participantes tiene que hacerse amiga de ellos, tiene que ser alguien en quien ellos sientan que pueden confiar, que no los va a delatar y que se preocupa por su seguridad y por su salud.
- Frente a la asequibilidad de los nuevos medicamentos aprobados y a su posterior comercialización ¿cuáles son los principales desafíos para América Latina?
NH. Hace unos años, 2015 y 2016 revisamos los medicamentos que la FDA había aprobado en 2012 que se habían testado en América Latina, eran 33, y nos encontramos con que un 30% no se habían registrado ni comercializado en ninguno de los países latinoamericanos donde se habían testado… Simplemente las empresas farmacéuticas no tenían interés en comercializar esos medicamentos en esos países y no los registraron. A veces los registraron, pero no los comercializaron porque esperaban a que las condiciones del mercado fueran más favorables [6].
Solo el 25% de los medicamentos (n=8) se comercializaron en todos los países en que se habían testado. Entre los medicamentos que se comercializaron, solo uno costaba menos de un salario mínimo mensual, la inmensa mayoría costaban más de cinco salarios mínimos mensuales – y en un caso hasta 899 veces.
Además, los boletines independientes de medicamentos habían estudiado la ventaja comparativa de 26 de estos medicamentos nuevos con los tratamientos existentes, 10 de ellos habían sido clasificados como No Usar, solo cinco de ellos se clasificaron como posiblemente superiores para poblaciones especiales, generalmente reducidas, y solo tres de ellos se habían comercializado en los países donde se habían testado. Es decir, la mayoría de los medicamentos que se testaron en América Latina, cuando se comercializaron, resultaron ser inasequibles para la mayoría de la población y de los sistemas de salud latinoamericanos, además la mayoría no son superiores a los tratamientos existentes [7].
- ¿Cuáles son las principales debilidades estructurales que ha identificado en los comités de ética en investigación clínica en América Latina, y cómo pueden fortalecerse para priorizar la salud pública sobre los intereses comerciales?
NH. Las debilidades que identificamos en América Latina no son específicas de la región… También están presentes en países de altos ingresos. El primer problema es que se puede constituir un CEI con solo 5 miembros, en la mayoría de los casos los CEI son más grandes, 7-15 personas, para garantizar quorum. Esos CEI revisan todo tipo de proyectos, desde ensayos clínicos con medicamentos financiados por la industria a tesis de estudiantes y ensayos con dispositivos médicos.
Además, suele ser una tarea mal remunerada y con frecuencia los miembros trabajan ad honorem. Como resultado, no todos los CEI que revisan protocolos de ensayos clínicos con medicamentos cuentan con miembros expertos en el área clínica en la que se va a aplicar el producto en investigación, ni con metodólogos especializados en ensayos clínicos. Es decir, si bien esos CEI pueden evaluar tesis de estudiantes, muchos de ellos no cuentan con la capacidad técnica necesaria para evaluar los diseños de los ensayos clínicos que les presenta la industria y que suelen estar muy bien escritos.
Los CEI tienen la posibilidad de consultar con expertos, pero pocos lo hacen.
Otro problema es que los ensayos clínicos son un negocio…. Eso es algo que no aparece en las formas de consentimiento informado pero que se discute si se debería incluir. Tanto los investigadores principales como los centros donde se realiza la investigación se benefician económicamente de los ensayos clínicos que realizan. Muchos de los CEI incluyen a miembros que hacen o han hecho ensayos clínicos financiados por la industria, y tienen un sesgo – no siempre reconocido- por favorecer a sus colegas… “Hoy por ti, mañana por mi” … También se sabe que a veces los administradores de los hospitales o centros de investigación imponen sus deseos de que se realicen ensayos clínicos.
Hay que resaltar que la mayoría de CEI no tienen acceso a los contratos entre el patrocinador y el investigador y/o el centro de investigación, por lo que no pueden evaluar si hay alguna cláusula del contrato que puede inducir a que se violen los criterios de inclusión en un ensayo clínico o a que se retenga a un paciente que debería retirarse del ensayo.
El resultado final es que los CEI suelen aprobar los ensayos clínicos que presenta la industria, lo que contrasta con las afirmaciones que han hecho metodólogos de renombre sobre la calidad del diseño de muchos de dichos ensayos clínicos.
Los países compiten en aprobar lo más rápido posible los ensayos de la industria, para conseguir más ensayos. Los miembros de los CEI que entrevistamos en diferentes países de América Latina (incluyendo Argentina, México, Costa Rica, Panamás, Perú y Colombia) dijeron que cuando evalúan ensayos clínicos financiados por la industria no solicitan cambios, porque si lo hacen se retrasaría el reclutamiento de pacientes y esto perjudicaría al investigador y a la institución [8].
En resumen, la mayoría de los CEI, no todos, suelen aprobar los ensayos clínicos financiados por la industria, la proporción de estudios que rechazan es muy baja y suele deberse a problemas administrativos.
Se puede pensar que los países/regiones tendrían que profesionalizar a los CEI que reciben los ensayos clínicos patrocinados, para que los evalúen expertos en la materia. Eso no eliminaría a los CEI institucionales, quienes podrían aceptar o no aceptar el dictamen del CEI especializado, y quienes se responsabilizarían de monitorear y proteger a los participantes.
- ¿Qué condiciones deberían establecerse para que Colombia se consolide como centro regional de ensayos clínicos sin comprometer la soberanía farmacéutica ni la protección de los participantes?
NH. Bueno, es que pienso que no se debería consolidar. No le veo las ventajas de hacerlo…
- ¿Cómo se beneficia Colombia y otros países de América Latina de realizar ensayos clínicos?
Agencias internacionales y países están afirmando que es importante que se hagan ensayos clínicos para: (1) dinamizar la economía; (2) desarrollar la capacidad de investigación. Sin embargo, no hemos encontrado evidencia para sustentar dichos argumentos.
Según la misma industria, en el 2016, por cada ensayo clínico que se hace en un país latinoamericano ingresan entre uno y dos millones de dólares… En el caso de Colombia esto representaría alrededor de US$140 millones (quizás el doble si se tienen en cuenta las externalidades) [9], pero eso no son ingresos netos, pues hay que descontar los gastos en los que incurre el INVIMA en la aprobación y supervisión de los ensayos clínicos, así como los gastos de los CEI.
También habría que tener en cuenta los gastos médicos de las personas que participan en ensayos clínicos y experimentan eventos adversos, que con frecuencia corren a cargo del erario público o de las aseguradoras, así como parte del tiempo del personal de salud que reclutan a los pacientes para los ensayos y tienen que verificar si cumplen los criterios de inclusión. Es decir, hay gastos asociados a la realización de los ensayos clínicos que en muchos casos no los pagan los patrocinadores, sino que corren a cargo del sector público o de las aseguradoras, o los absorbe el personal que dona su tiempo.
Que nosotros sepamos, nadie ha contabilizado lo que los países invierten para que la industria realice los ensayos clínicos, por lo que se desconoce cuál es el aporte neto de la industria de los ensayos clínicos a la economía colombiana o a la de cualquier otro país.
El otro argumento es que los ensayos clínicos contribuyen al desarrollo científico. Tenemos muchos problemas con esta afirmación, porque lo que los científicos deben aprender es a diseñar proyectos de investigación y analizar la información recabada… La gran mayoría de los que hacen ensayos clínicos patrocinados por la industria no participan de las discusiones sobre el diseño de la investigación ni del análisis de los datos, simplemente recaban datos… Eso no es capacitar en investigación nada más administran un proyecto diseñado en un país de altos ingresos, en donde también se analizarán los datos recabados en Colombia. En Colombia solo se recaban datos. Para mejorar las capacidades de investigación habría que invertir en becas de estudios doctorales, ya sea en Colombia o en el extranjero.
- Preguntamos al Dr. Bernardo Useche, ¿cómo las reflexiones hechas por la Dra. Homedes sobre los ensayos clínicos se pueden explicar en el contexto de la política farmacéutica en Colombia?
BU. La Doctora Homedes hizo un análisis en el que se revela información importante y poco conocida sobre los ensayos clínicos de las multinacionales farmacéuticas. Especialmente dos puntos llaman la atención:
- El hecho que son muy pocos los productos farmacéuticos, resultados de estos experimentos clínicos, que se aprueban y salen al mercado anualmente que realmente aportan algo nuevo al tratamiento de los pacientes con medicamentos ya existentes.
- Que estos ensayos clínicos son un excelente negocio en el que se benefician la industria, los centros de investigación, los investigadores y los profesionales o agencias que reclutan a los participantes, mientras poco o nada se benefician los pacientes.
El caso en Colombia de Carolina Jiménez en el cual, sin atender lo firmado en el consentimiento informado, no se informó a la participante de los resultados del ensayo clínico que comprometían en materia grave su salud, es ilustrativo [10].
En Colombia, cada vez son más los ensayos clínicos de la industria en busca de nuevos medicamentos que puedan patentar, dadas las ventajas que les otorgan los Tratados de libre Comercio con Europa y EE UU. Más aun, el gobierno de Gustavo Petro mediante reciente decreto presidencial mantendrá las normas de propiedad intelectual vigentes en los tratados de libre comercio [11].
Según el informe de la Asociación Colombiana de Centros de Investigación Clínica (ACIC) del 2024, 160 centros de investigación tenían en curso 332 ensayos clínicos a los que se adicionaron ese año otros 29 estudios por un valor de US$73 millones. De esos 361 experimentos, 89 ensayos clínicos fueron patrocinados por Merk Sharp & Dohme, 37 por Novartis, 27 por AstraZeneca y 19 por Bristol Myers [12].
Según la Asociación de Laboratorios Farmacéuticos de Investigación y Desarrollo (AFIDRO), a 28 de agosto 2025, ya son mil los estudios clínicos en Colombia [13].
Las multinacionales farmacéuticas, a través de AFIDRO, quieren avanzar y “consolidar a Colombia como un hub regional de investigación” en América Latina. En mi opinión, con esta estrategia, la industria tiene como objetivos ampliar su posición predominante en un mercado creciente de los adultos mayores, competir con China, país que se ha posicionado como potencia mundial en investigación clínica [14] y compensar la pérdida de rentabilidad que supone el control de precios impuesto en EE UU por el presidente Trump a través de varias de sus órdenes [15] presidenciales [16] ejecutivas [17].
Parte integral de esta estrategia de la industria es hacer lobby para que los gobiernos latinoamericanos aumenten el presupuesto de salud. Según la directora ejecutiva de la Asociación Colombiana de la Industria Farmacéutica (ASCIF), en Colombia, los medicamentos ya representan entre el 25 y el 30% de la Unidad de Pago por Capitación (Nota de SyF: UPC es el valor que el Estado colombiano paga por cada ciudadano, este valor es diferencial según ciclo de vida, según régimen de afiliación al sistema de seguridad social -régimen contributivo y subsidiado- y según área geográfica).
Si se tiene en cuenta que los productores de medicamentos patentados tienen como meta acceder a un mayor porcentaje de los recursos públicos, se puede prever que en la medida en que se haga realidad esta proyección, la sostenibilidad financiera del sistema de salud colombiano se verá seriamente afectada.
No hay duda de que las multinacionales deben continuar siendo un jugador clave en el mercado farmacéutico en Latinoamérica, pero se convierte en una necesidad fundamental fortalecer la industria nacional. Es un problema de seguridad y soberanía farmacéutica.
[1] Sidney M Wolfe. The seven-year rule for safer prescribing. Aust Prescr 2012;35:138-9 https://australianprescriber.tg.org.au/articles/the-seven-year-rule-for-safer-prescribing-1.html
[2] Elliott, C. Whatever Happened to Human Experimentation? Hastings Center Report, 2016;46: 8-11. https://doi.org/10.1002/hast.531
[3] Minaya G, Fuentes D, Ugalde A, Homedes N. A missing piece in clinical trial inspections in Latin America: interviews with research subjects in Peru. Journal of Empirical Research on Human Research Ethics; 2017;12(4) 232–245. https://pubmed.ncbi.nlm.nih.gov/28728496/
[4] ibid
[5] Ibid
[6] Homedes N, Ugalde A. Ensayos clínicos en América latina: implicancias para la sustentabilidad y seguridad de los mercados farmacéuticos y el bienestar de los sujetos. Salud Colectiva 2016; 12(3): 317-345 https://revistas.unla.edu.ar/saludcolectiva/article/view/1073
[7] Ibid
[8] Puede leer los resultados de estas entrevistas en https://www.saludyfarmacos.org/publicaciones/informes/
[9] Pugatch Consilium. Challenges and Opportunities -Developing the biotechnology sector in Colombia. 2016. https://www.pugatch-consilium.com/reports/Challenges%20and%20Opportunities_v6.pdf
[10] Palacios C. La Country me usó como conejillo. El Tiempo, 29 de agosto de 2025. https://www.eltiempo.com/opinion/columnistas/la-country-me-uso-como-conejillo-3485283
[11] Ministerio de Salud y Protección Social. Decreto Presidencial 858 de 2025, artículo 2.11.2.4.2. Mecanismos de participación para la investigación, desarrollo y producción nacional de Tecnologías Estratégicas en Salud. https://www.funcionpublica.gov.co/eva/gestornormativo/norma.php?i=261736
[12] Asociación Colombiana de Centros de Investigación Clínica (ACIC). Resumen de la Medición del aporte económico de la investigación clínica en Colombia, Estudio 2024. https://aciccolombia.org/wp-content/uploads/2025/03/INFOGRAFIA.pdf
[13] Asociación de Laboratorios Farmacéuticos de Investigación y Desarrollo (AFIDRO). La investigación clínica. Consultado 28-08-2025. https://afidro.org/la-investigacion-clinica/
[14] Bruckner T. New study shows that China is now a global powerhouse of clinical research. TranspariMed, 26 de mayo de 2025. https://www.transparimed.org/single-post/new-study-shows-that-china-is-now-a-global-powerhouse-of-clinical-research
[15] Presidency Executive Order 14273 of April 15, 2025; https://www.federalregister.gov/documents/2025/04/18/2025-06837/lowering-drug-prices-by-once-again-putting-americans-first
[16] Presidency Executive Order 14293 of May 5, 2025; https://www.federalregister.gov/documents/2025/05/08/2025-08267/regulatory-relief-to-promote-domestic-production-of-critical-medicines
[17] Presidency Executive Order 14297 of May 12, 2025; https://www.federalregister.gov/presidential-documents/executive-orders/donald-trump/2025

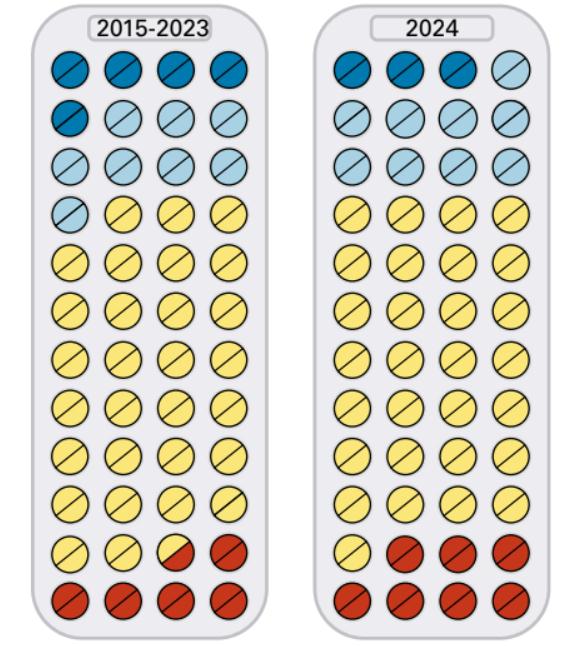
 Avance notable
Avance notable Avance mínimo
Avance mínimo Sin ventajas probadas
Sin ventajas probadas Más peligroso que beneficioso
Más peligroso que beneficioso





{kind=link}