Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Regulación, Registro y Diseminación de Resultados
A quién se aplica la Regla Común (Common Rule) de EE UU y qué significa para el consentimiento informado
Salud y Fármacos, 28 de abril de 2019
La información que resumimos a continuación proviene Outsourcing [1]
El 21 de enero de 2019 entró en vigor la Regla Común o el Common Rule que regula la investigación en la que participan seres humanos y que financia el gobierno federal de EE UU. En términos generales, cualquier institución que reciba fondos federales para nuevos estudios que incluyan sujetos humanos debe seguir la nueva guía, pero hay algunos matices.
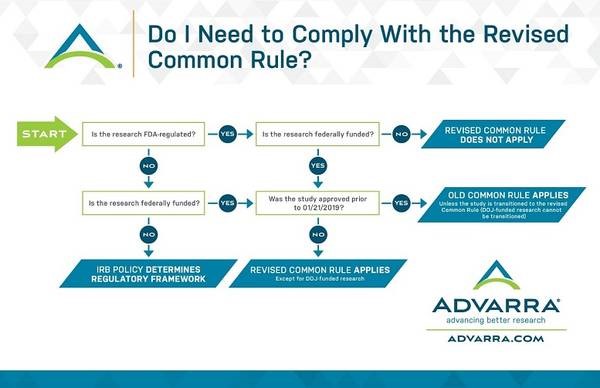
La Regla Común solo aplica a los estudios financiados por el gobierno federal que fueron aprobados después del 21 de enero de 2019. Los que fueron aprobados antes de esa fecha se rigen por la antigua Regla Común (Ver en inglés https://www.hhs.gov/ohrp/education-and-outreach/revised-common-rule/revised-common-rule-q-and-a/index.html). Si la investigación está financiada por el Departamento de Justicia, se tiene que aplicar la Regla Común independientemente de su fecha de aprobación. La nueva Regla Común no se aplica a la investigación regulada por la FDA. Sin embargo, los patrocinadores de la investigación regulada por la FDA podrían querer incluir cierta información clave y algunos elementos del consentimiento informado que aparecen en la nueva Regla Común.
Los cambios más importantes a la regla común tienen que ver con la ampliación de la definición de lo que se considera investigación en sujetos humanos, la revisión de las exenciones, la inclusión de nuevos elementos en el consentimiento informado, y el requisito de que el monitoreo de estudios de riesgo mínimo sea continuo. También establece que, si el estado no lo define, se puede seguir la política institucional para determinar quién podría ser un representante legalmente autorizado.
La FDA no ha revisado sus reglamentos para alinearlos con la Regla Común revisada. La investigación regulada por la FDA que no cuenta con fondos federales puede incorporar las disposiciones de consentimiento de la Regla Común revisada (información clave y los cuatro nuevos elementos de consentimiento). Las otras disposiciones (como las nuevas exenciones, los cambios en la revisión continua y los nuevos criterios de exención) no pueden aplicarse voluntariamente a la investigación regulada por la FDA. Los estudios aprobados antes del 21 de enero de 2019 también pueden agregar los cambios al contenido del consentimiento informado si así lo desean.
La Regla Común exige cambios en las formas de consentimiento informado, con el objetivo de que los sujetos de investigación tengan toda la información necesaria para decidir si quieren o no participar en el estudio. Ahora, las formas de consentimiento informado deberán incluir na sección nueva de información clave, que debe presentarse de manera organizada y concisa al comienzo de la forma de consentimiento informado (CI), y debe proporcionar la información que una persona razonable querría tener para tomar una decisión informada sobre si participar o no en el estudio. La práctica estándar es proporcionar la información que responda a las siguientes preguntas:
Si en esta sección de información clave se enumeran algunos de los elementos que hay que incluir en el CI (según 45 CFR 46.116 [b] y [c]), no hace falta repetir la información.
Los nuevos elementos que se deben incluir en la forma de consentimiento son (a. es obligatoria y el resto solo cuando sean apropiadas para el proyecto de investigación):
La información y / o muestras biológicas recopiladas como parte de esta investigación se pueden anonimizar para su uso en futuros estudios de investigación o se pueden distribuir a otro investigador para futuros estudios de investigación sin consentimiento informado adicional.
O
La información y / o muestras biológicas recopiladas como parte de esta investigación, aunque se eliminan los identificadores, no se utilizarán ni distribuirán para futuros estudios de investigación.
Es importante tener cuidado al implementar la segunda declaración en su forma de CI, ya que puede ser muy difícil mantener la información de los sujetos o las muestras biológicas para que no se utilicen en futuras investigaciones. Esto no quiere decir que la segunda declaración no se debe utilizar; solo tendrá que estar muy atento para asegurarse de que la información / muestras biológicas se distribuyan de acuerdo con el CI.
El esfuerzo por promulgar la nueva guía de la Regla Común ha sido un proceso largo y extenso. Seguramente quedan dudas que el Comité de Ética en Investigación debería poder aclarar.
Referencia