Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Una organización internacional sin ánimo de lucro para fomentar el acceso y el uso adecuado de medicamentos entre la población hispano-parlante
Agencias Reguladoras
EE UU
Cuando el medicamento enferma a los pacientes (When medicine makes patients sicker)
Kaiser Health News, 4 de enero de 2019
https://khn.org/news/how-tainted-drugs-reach-market-make-patients-sicker/
Traducido por Salud y Fármacos
A pesar del ruido rítmico, tipo martillo, de un ventilador mecánico, Alicia Moreno se había quedado dormida en una silla junto a la cama del hospital de su hijo de 1 año cuando un médico la despertó con malas noticias: el ablandador de heces de uso frecuente que unos meses antes le habían administrado a su hijo, Anderson, estaba contaminado con la bacteria Burkholderia cepacia.
De repente, la complicada evolución de Anderson tenía sentido médico. B. cepacia era la misma inusual bacteria que se encontraba misteriosamente en el tracto respiratorio del niño, y que lo había sacado temporalmente de la lista de candidatos a un trasplante de corazón. La misma bacteria reapareció después de su trasplante, se combinó con una enfermedad similar a la gripe e infectó sus pulmones. Desde entonces ha estado en un ventilador.
El medicamento contaminado de venta libre, docusato de sodio, que se receta rutinariamente a casi todos los pacientes hospitalizados para evitar el estreñimiento, hizo que Anderson sufriera “lesiones graves y peligrosas que amenazan su vida”, afirma una demanda presentada por su familia. Eventualmente, el medicamento fue retirado, pero solo después de que el personal de un hospital de Texas notara un aumento en las infecciones por B. cepacia, se inició una investigación de seis meses que desembocó nuevamente en el medicamento contaminado y su planta de fabricación en Florida.
“Algo que se suponía que lo ayudaría lo perjudicó”, dijo Alicia Moreno.
Desde el inicio de 2013, las compañías farmacéuticas con sede en EE UU o en el extranjero han retirado del mercado alrededor de 8.000 medicamentos, que representan miles de millones de tabletas, frascos y viales que han ingresado a la cadena de suministro de medicamentos en EE UU y se han colocado en los botiquines de medicamentos de los pacientes, en los armarios de suministros hospitalarios y en los goteos intravenosos, muestra una investigación de Kaiser Health News. Los retiros de mercado representan una fracción de los medicamentos que se envían anualmente. Pero los productos defectuosos contenían de todo, desde bacterias peligrosas o pequeñas partículas de vidrio hasta moho, o demasiado o muy poco ingrediente activo del medicamento.
Durante el mismo período, 65 instalaciones de fabricación de medicamentos retiraron casi 300 productos durante los 12 meses posteriores a haber superado una inspección de la FDA, incluyendo el caso del ablandador de heces, según un análisis de KHN de los avisos de retiro y los registros de inspección de la FDA.
Estos retiros incluyeron más de 39.000 botellas del medicamento contra el VIH Atripla por estar contaminados con “partículas de goma de silicona roja”, casi 37.000 tabletas genéricas de Abilify que eran “superpotentes” y casi 12.000 cajas de Aleve genérico (naproxeno) que en realidad eran ibuprofeno, según los datos de retiros analizados por KHN.
Según los informes de la FDA y los CDC, el medicamento que presuntamente enfermó a Anderson Moreno infectó gravemente a al menos otras 63 personas en 12 estados. El medicamento se fabricó en una planta de PharmaTech en el condado de Broward, Florida. Esa misma planta pasó una inspección de la FDA mientras estaba fabricando productos contaminados con bacterias, según una revisión de KHN de los registros de inspección.
Retiro de medicamentos después de inspecciones de la FDA
Cerca de la mitad de los medicamentos retirados había sido producidos en plantas que la FDA ya había identificadas por violaciones durante su última visita de inspección. Pero casi 700 retiros tuvieron lugar en plantas que había aprobadas después de una inspección. Y casi 3.000 retiros fueron de plantas que no se habían inspeccionado desde 2008 (véase el cuadro adjunto)

PharmaTech no respondió a las solicitudes de comentarios de KHN. Un abogado de la farmacéutica presentó una moción para desestimar la demanda en abril, pero no fue concedida. En los registros judiciales de seguimiento, PharmaTech ha negado las acusaciones en su contra.
Al igual que otros comisionados de la FDA antes que él, Scott Gottlieb ha tildado al programa de supervisión de medicamentos de su agencia como “estándar de oro” por su seguridad y eficacia.
Pero el consultor veterano de la industria John Avellanet, quien ha capacitado a inspectores de la FDA, cuestiona la efectividad de las inspecciones que la FDA hace a las plantas de medicamentos. “Es muy fácil” para los inspectores de la FDA ignorar cosas porque están trabajando con términos normativos confusos.
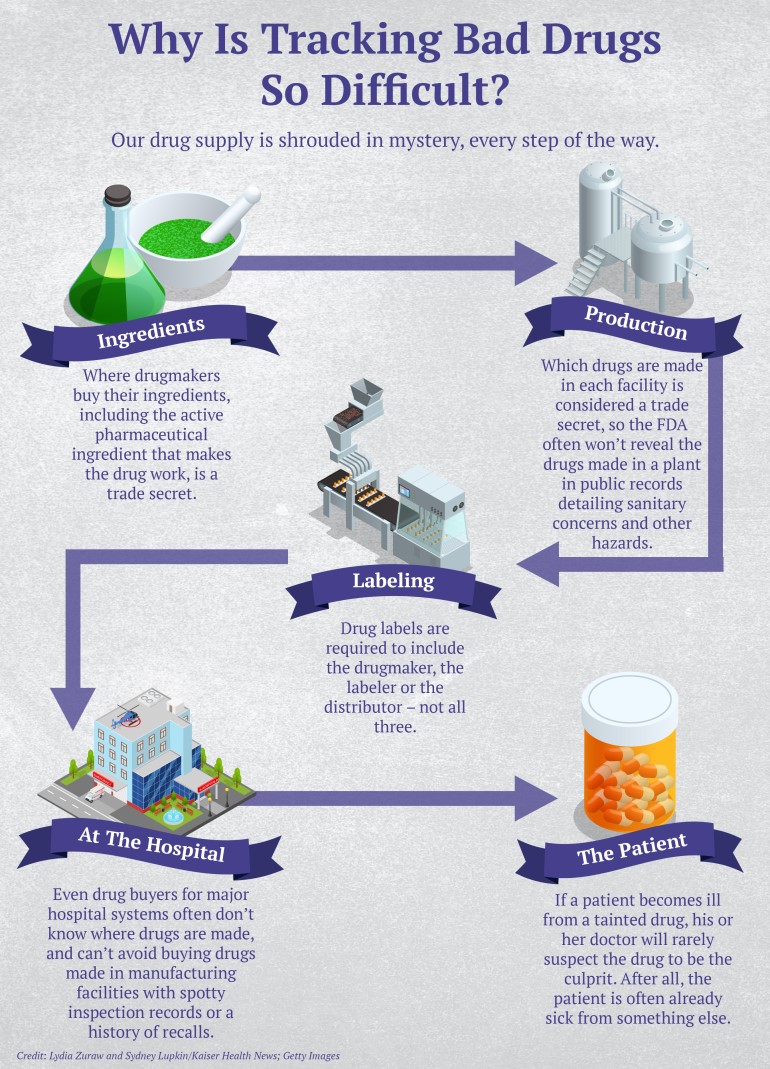
Es casi imposible determinar con qué frecuencia las personas se enferman o mueren a causa de medicamentos contaminados. Ninguna agencia gubernamental rastrea los casos a menos que estén vinculados a un brote importante entre pacientes hospitalarios. Y cuando surgen enfermedades repentinas y aparentemente aleatorias en lugares dispares también son notoriamente difíciles de relacionar con un medicamento contaminado. Eso es en parte porque los fabricantes de medicamentos no tienen que divulgar qué productos se fabrican en qué plantas de fabricación, ya que se considera información de propiedad exclusiva.
El resultado: incluso el que compra medicamentos para un hospital importante no puede rastrear de dónde proviene un producto potencialmente peligroso, dijo Erin Fox, quien compra medicamentos para los hospitales de la Universidad de Utah Health.
“La seguridad del paciente debe ser lo primero”, dijo, y agregó que el análisis de KHN indica que “la calidad de nuestro medicamento probablemente no es la que creemos”, y lo califica de una realidad “aterradora”. “Algo debe cambiar si esto sucede con tanta frecuencia y tenemos pacientes que reciben productos contaminados”.
La FDA rechazó ser entrevistada para esta historia, pero respondió las preguntas escritas.
“Si bien la FDA preferiría que no se distribuyeran medicamentos que luego se van a retirar, no creemos que un retiro indique el fracaso de los programas de inspección y vigilancia de la FDA”, dijo el portavoz de la FDA, Jeremy Kahn, en un correo electrónico. Dijo que los inspectores “pueden no descubrir todos los problemas o prácticas que pueden llegar a provocar un problema que ocasione un retiro” y que “no todos los retiros son el resultado de una mala práctica de fabricación”.
La historia de PharmaTech
“Aleta afortunada, aleta afortunada, aleta afortunada”, Alicia Moreno, de 30 años, repetía mientras desenredaba el brazo de su hijo de 3 años debilitado por un accidente cerebrovascular de un suéter y su ventilador portátil en el asiento trasero del automóvil para emprender un viaje de cuatro horas para visitar a los médicos en Ann Arbor, Michigan. En la película de Disney “Buscando a Nemo”, el padre de Nemo dice que la aleta más pequeña del joven es su “aleta afortunada”.
Mientras su marido conduce, Alicia saca una caja de plástico transparente con jeringas y mira el reloj en el tablero. Anderson necesita alrededor de dos docenas de medicamentos diferentes cada 24 horas, y Alicia se los administra a través de la palomilla que tiene en su barriga a las horas designadas.
No siempre fue así. Anderson parecía estar sano hasta su chequeo de 6 meses en mayo de 2016, dijo su madre. A mitad del examen, los Moreno llevaron a su bebé a un hospital cercano y se enteraron de que tenía una insuficiencia cardíaca y necesitaría un trasplante para sobrevivir. Ahí es donde recibió el ablandador de heces contaminado, alegan sus abogados. El hospital donde Anderson finalmente recibió su trasplante confirmó por correo electrónico que Anderson dio positivo a la misma cepa de B. cepacia involucrada en el brote que se encontró en el medicamento contaminado de PharmaTech.
En julio, según la familia, Anderson comenzó a tener dificultad para respirar y su temperatura subió a 106 grados, por lo que ingresó en cuidados intensivos, donde los médicos y las enfermeras lo cubrieron de hielo y se apresuraron a buscar la causa. Sus pruebas dieron positivo para B. cepacia, una bacteria que se encuentra en el agua no tratada y que por lo general no enferma a las personas sanas. La situación de Anderson en la lista de trasplantes quedó en suspenso y su problema cardíaco empeoró. Lo colocaron en una máquina que transfirió sangre fuera de su cuerpo, la oxigenó y la devolvió nuevamente a su cuerpo.
Anderson finalmente recibió un trasplante de corazón en noviembre de 2016, pero cuatro días después de que los médicos cerraran su pecho, volvió a tener fiebre y sus pulmones empeoraron, por lo que requirió una maquinaria más complicada. Las pruebas dieron positivo para un virus similar a la gripe y a B. cepacia, según el hospital.
“¿De dónde lo sacó?”, suplicaban sus padres. En ese momento, nadie lo sabía.
Cómo se deslizan las drogas contaminadas a través de descuidos
Se supone que la FDA inspecciona todas las fábricas, extranjeras y nacionales, que producen medicamentos para el mercado de EE UU, pero una revisión de KHN de miles de documentos de la FDA (registros de inspección, retiros del mercado, cartas de advertencia y juicios) ofrece información sobre las formas en que los medicamentos mal fabricados o contaminados llegan a los consumidores: los inspectores no detectan riesgos graves. Los fabricantes de medicamentos no cumplen con los estándares ni siquiera cuando la FDA toma medidas para forzar su cumplimiento. Cientos de plantas no han sido inspeccionadas durante años, si es que alguna vez lo han sido.
En julio pasado, por ejemplo, la FDA anunció el primero de muchos retiros voluntarios del medicamento valsartán para la presión arterial, porque algunas tabletas contienen una impureza que causa cáncer y se llama N-nitrosodimetilamina (NDMA). Más tarde encontrarían un carcinógeno similar, N-nitrosodietilamina (NDEA), en las píldoras de valsartán. Durante los dos años anteriores, los investigadores detectaron problemas preocupantes en dos fábricas extranjeras involucradas en la fabricación del medicamento.
Los investigadores de la FDA encontraron óxido, pintura desconchada y equipos deteriorados en una planta gestionada por Zhejiang Huahai Pharmaceutical Co. en Zhejiang, China. El personal de la planta no estaba testando e investigando correctamente las “anomalías” en sus medicamentos y descartaban resultados de pruebas problemáticas, dijo la FDA en ese momento. Los inspectores también encontraron “partículas metálicas negras” y otros problemas en algunos medicamentos no identificados.
La FDA inspeccionó la planta en julio de 2018, después de recibir quejas por NDMA procedentes de una instalación que estaba más abajo en la cadena de suministro de medicamentos. La FDA puso la instalación en alerta de importación a finales de septiembre y emitió una carta de advertencia en noviembre detallando las deficiencias, incluyendo “Fallos de su unidad de calidad para garantizar que las quejas relacionadas con la calidad se investiguen y resuelvan”.
En una instalación de Hetero Labs en India, en 2016, los inspectores de la FDA encontraron residuos de colores y blancos en los componentes, algunas tabletas de fábrica tenían el doble de grosor que otras, y los empleados estaban destruyendo documentos a media noche. La FDA emitió una carta de advertencia a la compañía como resultado de la inspección.
Las plantas que fabrican medicamentos para los consumidores estadounidenses se deben inspeccionar cada pocos años, según un sistema basado en riesgo. Sin embargo, en la última década, la FDA no ha inspeccionado la calidad de los medicamentos de más de 2.500 instalaciones en más de cinco años, según un análisis de KHN. Según el estudio, la FDA no tiene informes de haber inspeccionado de calidad de los medicamentos de más de 1.200 plantas domésticas y cerca de 400 plantas extranjeras en la última década, excluyendo aquellas que fabrican productos para animales. Gottlieb dijo en diciembre que espera eliminar la acumulación de instalaciones de medicamentos no inspeccionadas para fines de septiembre de 2019.
En el mejor de los casos, las inspecciones son una imagen instantánea en el tiempo e implica analizar los procesos en lugar de evaluar los medicamentos en sí mismos, dijo el especialista en calidad de medicamentos Dinesh Thakur, quien ha trabajado para los fabricantes de medicamentos. Las inspecciones pueden llevarse a cabo mientras la instalación está produciendo solo uno de la docena de medicamentos que generalmente fabrica.
“El supuesto implícito … es que si los procesos [de fabricación] son sólidos, el producto será de buena calidad”, dijo Thakur, quien dio la alarma sobre los problemas de control de calidad del fabricante de medicamentos genéricos Ranbaxy, que resultó en una declaración de culpabilidad en 2013 y un acuerdo de US$500 millones. “Sus datos nos dicen que esto no es cierto”.
Muchas de las inspecciones, dijo, están “se gestionan por etapas”, por lo que las fábricas superan la prueba el día señalado, pero “cuando los inspectores se van, es una historia completamente diferente”.
David Gortler, un exfuncionario médico de la FDA dijo que la mayoría de las inspecciones de plantas de medicamentos implican revisar los registros en papel y confiar en que son reales, en lugar de probar medicamentos al azar.
“Cualquiera puede escribir cualquier cosa en un pedazo de papel”, dijo Gortler, quien ahora es consultor en FormerFDA.com. Añadió que los inspectores de la FDA no son amonestados, ni siquiera informados, de que han dado el visto bueno a una planta que poco después emitió un retiro del mercado.
Un golpe de suerte resuelve un misterio
El ablandador de heces contaminado que presuntamente enfermó a Anderson Moreno fue uno de los muchos medicamentos retirados por plantas poco después de que pasaron una inspección de la FDA. La bacteria se detectó solo después de un brote de la enfermedad, y después de una buena investigación médica.
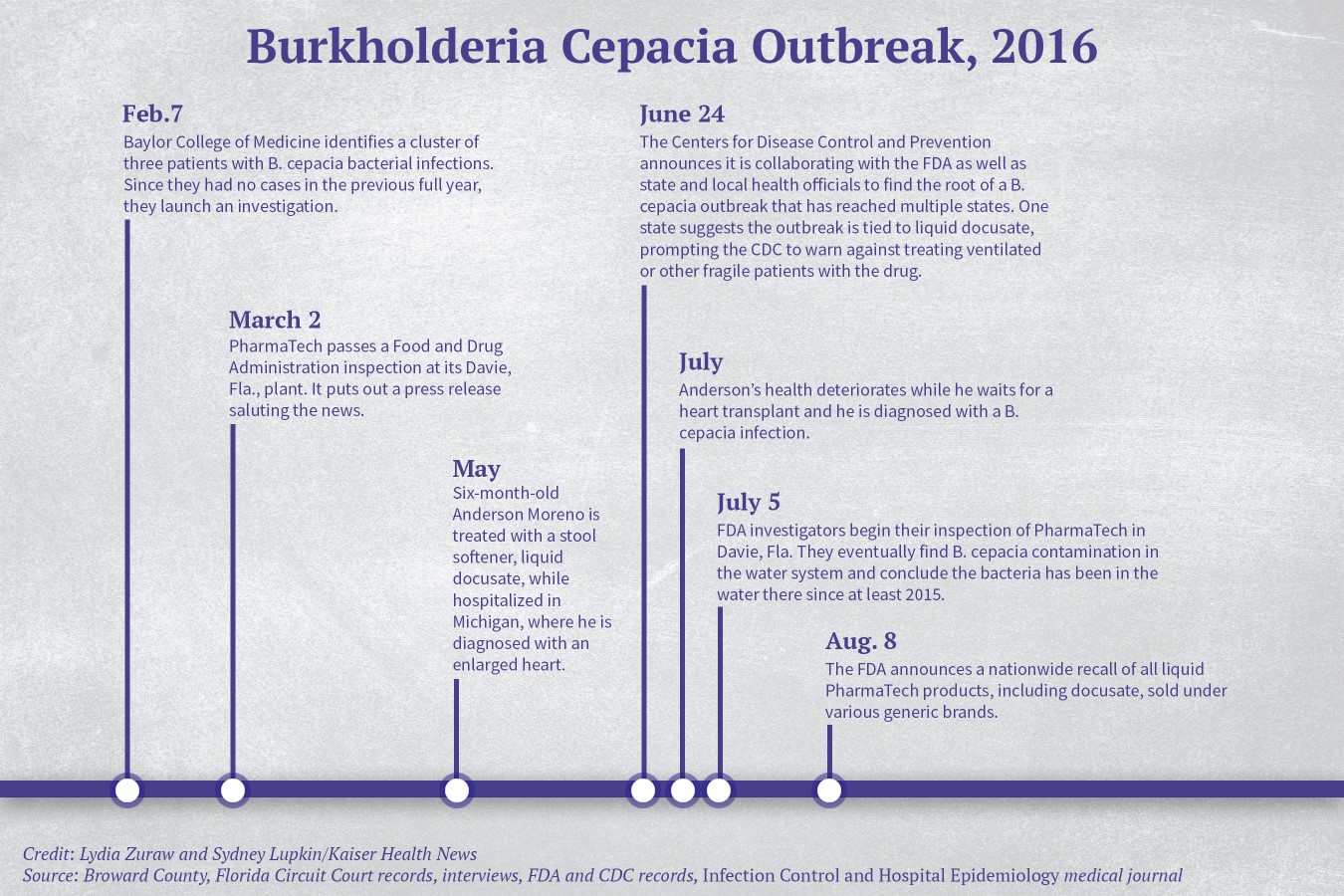
En febrero de 2016, a más de 1.000 millas de distancia de la cama de Anderson en la UCI en Michigan, el personal de la UCI pediátrica del Texas Children’s Hospital en Houston diagnosticó tres casos de B. cepacia en una semana, según un artículo de 2017 publicado en Infection Control and Hospital Epidemiology. Era extraño porque no había habido casos el año anterior.
El personal del hospital se embarcó en una investigación que duró meses y en julio habían identificado a 24 víctimas, cuya mediana de edad era inferior a 2 años. Los pacientes tenían la misma cepa de bacterias en su sangre, sus vías respiratorias, su orina o sus heces, según el artículo.
Las muestras coincidieron con las bacterias encontradas en el docusato líquido, el ablandador de heces, escribieron los investigadores.
El hospital alertó a los CDC y otros funcionarios de salud pública de sus hallazgos. Los CDC identificarían finalmente 63 infecciones confirmadas y 45 sospechas de infección grave por B. cepacia vinculados a la droga contaminada en 12 estados.
Una inspección de 36 días de la FDA a PharmaTech en Davie, Florida, que finalizó el 9 de agosto de 2016, reveló que la bacteria estaba en el agua utilizada para limpiar equipos y fabricar productos líquidos. Los inspectores de la FDA concluyeron que la bacteria entró en los medicamentos de la instalación en el 2015 y todavía estaba presente en el agua.
Anderson recibió tratamiento con el ablandador de heces en mayo de 2016. Sus padres presentaron una demanda en septiembre de 2017 en el condado de Broward (Florida), donde se ubica PharmaTech, contra el fabricante de medicamentos y otros en la cadena de suministro de medicamentos, alegando que la droga estaba contaminada y causó graves daños a su hijo. PharmaTech, que no devolvió las solicitudes de comentarios de KHN, presentó una moción para desestimar el caso, pero sin éxito, y en presentaciones posteriores ha rechazado todos los cargos.
Brote de B cepacia, 2016
En Pittsburgh, una niña de 9 meses de edad que había recibido el ablandador de heces murió el 4 de mayo de 2016, según una demanda presentada por su familia en julio de 2017 en el Tribunal de Distrito de EE UU para el Distrito Oeste de Pennsylvania. Su madre se enteró del retiro del medicamento por casualidad y preguntó al hospital si su hija fallecida había recibido el medicamento contaminado, dijo su abogado a KHN. La familia presentó cargos contra PharmaTech y otros en la cadena de suministro de medicamentos en una demanda por homicidio culposo. El tribunal rechazó las mociones de PharmaTech para desestimar el juicio, y el fabricante de medicamentos en una presentación posterior negó toda responsabilidad. En noviembre de 2017, un abogado que representa a PharmaTech en ese caso de muerte injusta dijo al Sun Sentinel de Orlando (Florida) que se defenderá contra las acusaciones y que no podía hacer más comentarios “debido a la naturaleza del litigio en curso”.
Según los registros federales, los inspectores de la FDA tuvieron la oportunidad de detectar la contaminación durante su inspección de marzo de 2016, pero la planta de PharmaTech pasó la inspección sin citaciones. El gerente general de PharmaTech, Ray Figueroa, resaltó los resultados de la inspección en un comunicado de prensa, calificándolos de “un testimonio del compromiso de PharmaTech para la fabricación de productos de la más alta calidad”.
Cómo pueden salir mal las cosas
A lo largo de los años, la FDA ha emitido miles de reprimendas contra las plantas de medicamentos, citando violaciones de seguridad, emitiendo cartas de advertencia y bloqueando las importaciones de ciertas plantas extranjeras. En casos raros, la FDA también puede incautar medicamentos y lo ha hecho 23 veces en la última década. La última incautación de medicamentos fue hace más de dos años, según los registros de la FDA.
En un comunicado enviado por correo electrónico, el comisionado de la FDA, Gottlieb, dijo que la FDA está “adoptando nuevas iniciativas” para identificar los problemas antes de que ocurran y que “no va a ser tímida” al usar sus poderes para mitigar los riesgos.
Pero el sistema puede ser bloqueado o distorsionado, y las capacidades de la FDA para forzar el cumplimiento son limitadas. Por ejemplo, no tiene el poder de obligar a que se retire un medicamento, cuando se llama la atención por problemas de fabricación no impone multas.
Muchos casos salen a la luz solo cuando un informante hace sonar una alarma.
Thakur, el informante de Ranbaxy, dijo que los funcionarios de otros países a veces informan a las plantas sobre las inspecciones “sorpresa” de la FDA. Y los inspectores de la FDA a menudo tienen que confiar en traductores contratados por las compañías farmacéuticas, dijo Avellanet, quien ha sido consultor de inspecciones de plantas de medicamentos durante más de 20 años.
En Nippon Fine Chemical, Japón, los empleados se pararon “hombro con hombro” para impedir la entrada de un funcionario de la FDA en diciembre de 2015, según una carta de cumplimiento enviada a la planta y publicada en línea.
Menos de un año después, en India, presuntamente Vikshara Trading & Investments Ltd. fingió una huelga trabajadores para bloquear la entrada a la planta, según un documento de la FDA que describía las “declaraciones falsas” del fabricante. Cuando finalmente se permitió la entrada de inspectores, mantuvieron apagadas las luces.
“Nuestro investigador tuvo que realizar partes del recorrido en la oscuridad, usando una linterna”, se lee en la carta de advertencia de la FDA, y agrega que un polvo no identificado estaba “esparcido” y “apelmazado en el piso” de las áreas de producción y se había detectado en productos farmacéuticos terminados.
Dos ex empleados presentaron una demanda contra Gilead Sciences, alegando que mintió a la FDA sobre el uso de una planta de fabricación de medicamentos en Corea del Sur, cuando en realidad estaba utilizando una instalación no registrada en China. Según la demanda civil presentada en septiembre de 2014 en el Tribunal de Distrito de EE UU para el Distrito Norte de California, el ingrediente producido en Synthetics China y utilizado en los medicamentos contra el VIH, Truvada y Atripla, contenía “fragmentos parecidos a vidrio”, “partículas negras parecidas al caucho” “partículas similares a plástico”, “partículas pequeñas como piedras o guijarros” y “fragmentos de metal”.
Los denunciantes alegaron que la planta de Gilead en Alberta, Canadá, tenía la tarea de tamizar contaminantes y ayudar a ocultar dónde se producía el ingrediente.
Dijeron que un lote del ingrediente estaba contaminado con arsénico, cromo y níquel. Otro tenía una bacteria peligrosa llamada Bacillus cereus, según los informantes. Aun así, Gilead lanzó el producto y no inició una retirada, alegaron los informantes.
En 2014, años después de que los informantes dejaran de trabajar para Gilead, el fabricante de medicamentos emitió dos retiros voluntarios de medicamentos contra el VIH, con aproximadamente siete meses de diferencia. Ambos recuerdan la contaminación que hemos mencionado con partículas de caucho de silicona roja.

Gilead declinó hacer comentarios. Gilead se ha opuesto a la demanda, alegando que, dado que el gobierno conocía las acusaciones y no las penalizaba negando las aprobaciones o con multas, la demanda no podía avanzar. En 2015, un juez federal desestimó el caso, pero un panel de la Corte de Apelaciones del 9º Distrito revocó esa decisión en 2017. Ahora la Corte Suprema puede escucharla; en abril de 2018 invitó al procurador general a presentar un escrito, “expresando los puntos de vista de EE UU”. El Departamento de Justicia presentó un informe en noviembre, diciendo que continuar con la demanda “no es de interés público”.
Dado que la FDA tiene poco poder para obligar a un fabricante de medicamentos a solucionar problemas o a retirar productos del mercado, los inspectores de la FDA a menudo señalan las mismas violaciones una y otra vez. Un análisis de KHN encontró que, durante la última década, 70 plantas de medicamentos, la mayoría de ellas domésticas, fueron penalizadas por la misma violación al menos cuatro veces. Y más de un tercio de esas plantas han emitido un retiro en algún momento.
Altaire Pharmaceuticals en Nueva York ha recibido citaciones de inspectores de la FDA cinco veces por “procedimientos inadecuados para productos farmacéuticos estériles”. En 2013, retiró 363.746 botellas de gotas genéricas para los ojos que se vendían en CVS, Target y Walmart por problemas de esterilidad, especialmente moho, porque el conservante del producto “puede no ser efectivo” hasta la fecha de caducidad. En general, a Altaire se le dijo que corrigiera 15 violaciones al menos dos veces.
KHN intentó ponerse en contacto con Altaire Pharmaceuticals, pero la compañía no respondió
Comprando a ciegas
Aproximadamente un año después de la retirada inicial de PharmaTech en 2016, la FDA anunció otra retirada de los mismos medicamentos por la misma bacteria: B. cepacia. Cuando Erin Fox vio el segundo retiro, pensó que era un error. La alerta decía que se evitaran todos los medicamentos fabricados por PharmaTech bajo varias etiquetas “y posiblemente [los productos de] otras compañías”. ¿Qué otras compañías? Fox se preguntó ¿Cómo podrían no saber cuáles?
Los médicos del hospital pidieron a Fox que retirara todos los productos hechos por PharmaTech de los estantes, pero debido a que las leyes de etiquetado son laxas, dijo, no podía estar segura de cuáles eran. Las etiquetas de los medicamentos deben incluir solo al fabricante, al empacador o al distribuidor, no a los tres, por lo que los médicos sugirieron que llamara a PharmaTech y preguntara qué más fabrica y para quién.
“Por supuesto”, dijo Fox, “no nos lo dijeron”.
Metodología
Para analizar las inspecciones y retiros de plantas que fabrican medicamentos, KHN comenzó con dos bases de datos de retiros de medicamentos de la FDA: una en OpenFDA y otra en el panel de datos de retiros de la FDA. La primera proporcionó detalles sobre los medicamentos, las fechas y las cantidades retiradas del mercado, y la segunda ayudó a identificar la planta que causó el retiro, denominada FEI. Los usamos para crear una base de datos de retiros más completa.
Las FEI sirvieron como puente entre los datos de retiros y dos cuadros de datos de inspección. Ambos cuadros contenían fechas y propósitos de la inspección, pero uno enumeraba el puntaje de la inspección y la otra contenía una lista de citaciones. La combinación de las bases de datos de inspección y retiradas nos permitió encontrar la inspección más reciente de cada planta que precedió a una retirada y determinar su puntaje. También nos permitió contar las citaciones repetidas y determinar si las plantas que las recibieron alguna vez iniciaron un retiro.
Para determinar si las plantas no se habían inspeccionado en la última década, comparamos nuestros datos de inspecciones con la base de datos del registro actual de establecimientos de medicamentos (https://www.accessdata.fda.gov/scripts/cder/drls/default.cfm), que contiene todas las plantas operativas registradas. Excluimos las plantas que según la base de datos fabricaban productos para animales y aquellas que no “fabricaban” medicamentos explícitamente. La FDA ha dicho que puede haber una demora en agregar inspecciones a su base de datos una vez se han hecho.
Nuestros datos están actualizados a principios de octubre de 2018. En el análisis incluimos solo las inspecciones categorizadas como inspecciones de “garantía de calidad de los medicamentos”.